In this paper, one imidazole macrocyclic divalent cation with flexible configuration was chosen with the aim of the immobilization of its conformation. One novel organic-inorganic hybrid supramolecule {[syn/syn–OM–IM–C] (CdI2)[syn/anti–OM–IM–C’](Cd’I’2)} (1), [OM–IM–C2+ = (12Z,52Z)–11H,51H–1,5(1,3)-diimidazol-3-iuma-3,7(1,2)-dibenzenacyclooctaphane-13,53-diium] has been synthesized through the self-assembly reaction of OM–IM–C2+ with CdI2 under solvothermal reaction. This compound has been unambiguously confirmed by PXRD, IR, TG, Elemental analysis, theoretical calculation and X-ray single crystal diffraction. Crystallographic analysis shows that the anion of compound 1 is mononuclear structure. The stability of compound 1 is relatively good.
Introduction
Most of the anion receptors are based on pyridine [1, 2], imidazoles [3] and so on as hydrogen bond donors in their structure as anion binding sites [4]. The most representative of the macrocyclic templates is the imidazolium ring. The positive charge of imidazolium can not only be used as a host molecule building unit, but also can be used as a special guest molecule to form a self-assembled form of pseudo-rotaxane with a host molecule such as cucurbituril. In the field of catalysis, imidazolium cations are the precursor of nitrogen-hybrid carbene, and the resulting nitrogen-hybrid carbene metal organic compounds can be efficiently applied to homogeneous catalytic reactions. But there have been few reports on the synthesis of supramolecular compounds by self-assembly of metal halides or pseudohalogens using imidazolium macrocyclic cations as organic templating agents.
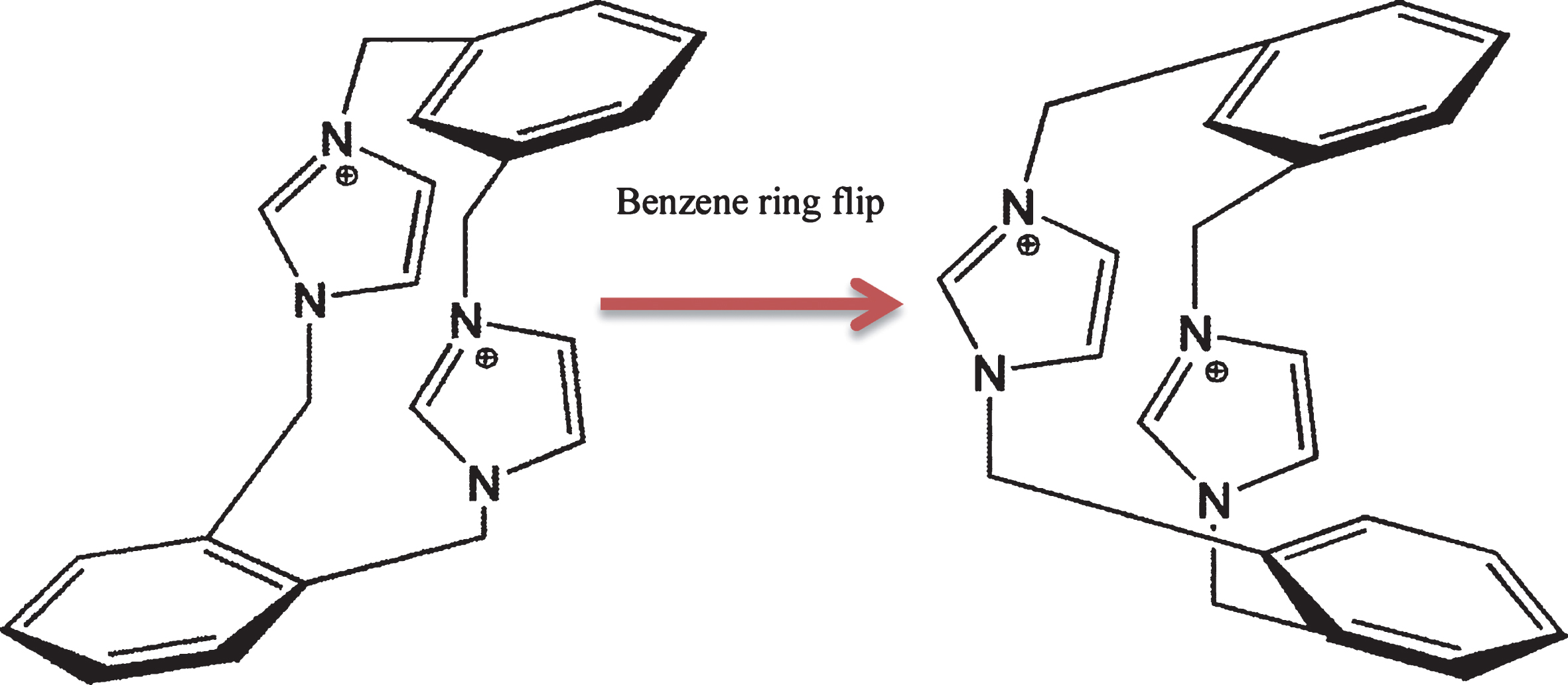
In this paper, the ortho-biimidazolium ring [5–19] was selected for the first time as a target model for conformational solidification and one kind of imidazolium-based macrocyclic supramolecular compound was synthesized by self-assembly. Compound 1 contain the identified conformation of syn/syn–OM–IM–C2+ and syn/anti–OM–IM–C2+. Imidazolium rings are mutually syn, which shows one syn-conformation. Flip of the benzene rings, which showed anti- and syn- conformation. Syn/anti and syn/syn conformation show in Scheme 1.
Syn/anti and syn/syn conformations of organic cation OM–IM–C2+ in compound 1.
Experimental
Materials and methods
The organic cation template (12Z,52Z)–11H,51H–1,5(1,3)-diimidazol-3-iuma-3,7(1,2)-dibenzenacyclooctaphane-13,53-diium (OM–IM–C·2PF6) was synthesized according to the reported procedure [5–19]. The IR spectra were measured on a Shimazu IR 435 spectrometer adopting KBr pellets in the scale of 400–4000 cm–1. Element analyses of C, H and N were performed using a Perkin-Elmer 240 elemental analyzer. Powder XRD pattern was collected on a Philips X-pert X-ray diffractometer at a scanning rate of 4°min–1 in the 2θ range from 6 to 53° with graphite monochromatized Cu-Kα radiation (λ= 0.15418 nm) with an X’ Celerator detector.
Supramolecular syntheses and conformation immobilization
Synthesis of [syn/syn–OM–IM–C](CdI2)[syn/anti–OM–IM–C’](Cd’I’2) (1)
Compound 1 was prepared by solvothermal synthesis method. For compound 1 1.5 mL CH3CN solution of OM–IM–C·2PF6 (0.01 g) was added into CdI2 (0.01 g) dissolved in 0.5 mL CH3CN with excess KI. The mixture was constantly stirred and reacted at 80°C for 72 hours and then cooled to room temperature within 14 hours. Colorless transparent needle-shaped crystals of 1 were obtained with a yield of 30%. IR (KBr, cm–1): 3443.88(m), 3121.64(w), 2924.21(w), 1550.12(w), 1449.18(w), 1144.79(s), 832.11(s), 747.96(w), 559.59(s), 485.44(w), Anal. Calc: C, 27.45; H, 2.3; N, 5.82; Found: C, 27.44; H, 2.38; N, 5.81%.
X-ray crystallography study
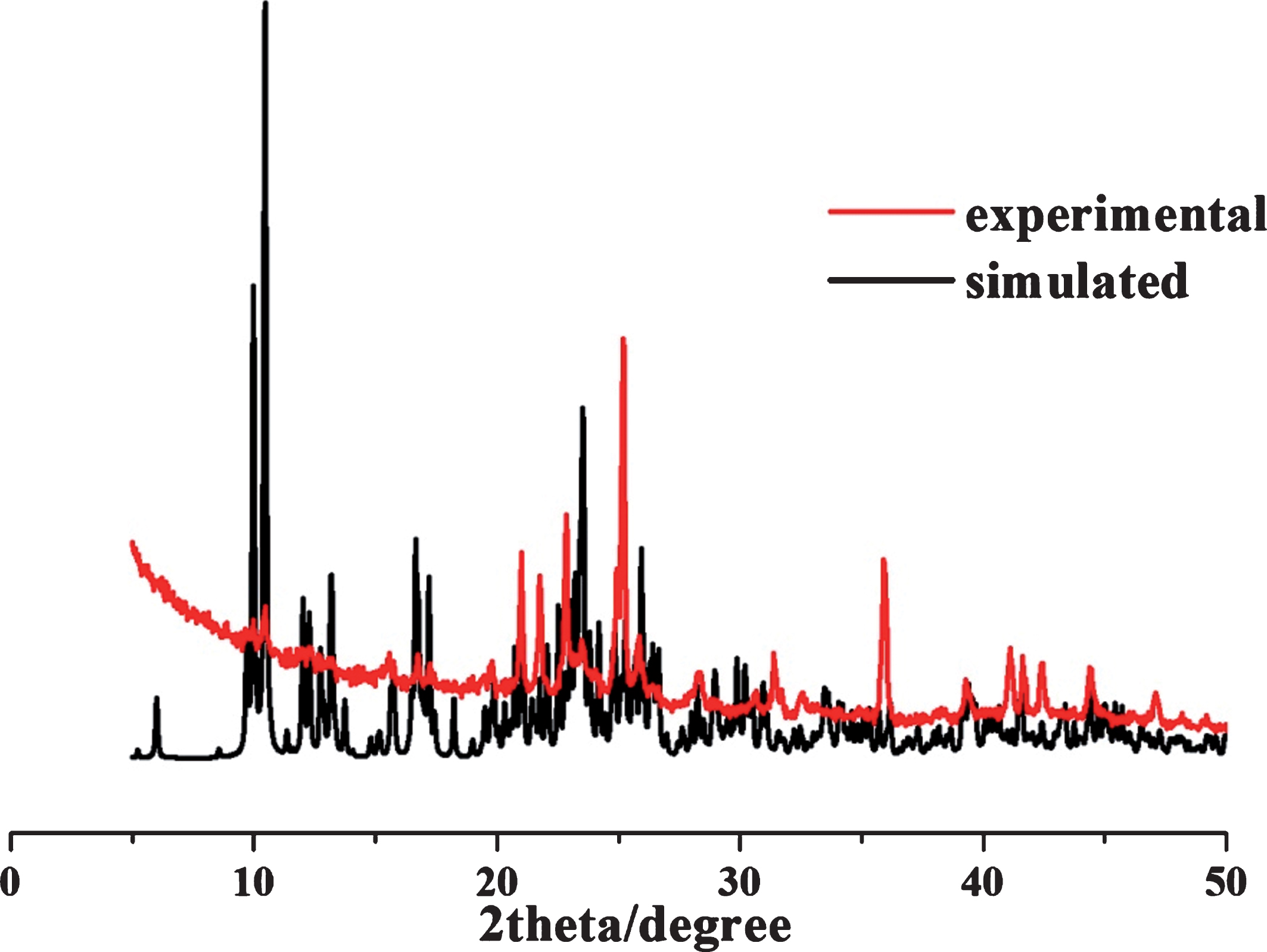
The X-ray single crystal diffraction data of compound 1 was recorded on the Bruckner SMART CCD diffractometer with graphite-monochromatic Cu-Kα radiation (λ= 0.71073 Å) at 293 K. Data reduction and absorption correction were done using the SADABS software package. After absorption correction, employ SHELXTL-97, OLEX-2 and other packages for analysis. The main bond lengths of the crystallographic data of compound 1 are shown in Tables 1 and 2. The purity of compound 1 was studied by X-ray powder diffraction (PXRD). For compound 1, the as-synthesized PXRD patterns closely match the simulated patterns generated from the result of the single-crystal diffraction data, indicative of pure products (Fig. 1). Simulation of the PXRD spectra was carried out by the single-crystal data and diffraction-crystal module of the Mercury (Hg) program available free of charge via the internet at http://www.iucr.org.
Crystal data and structure refinement details for 1
Compound
1
Empirical formula
C22H22CdI4N4
Formula weight
962.43
Temperature
293(2)
Crystal system
triclinic
Space group
P–1
a/Å
9.2511(3)
b/Å
18.1144(8)
c/Å
18.1818(10)
α/°
69.201(5)
β/°
80.619(4)
γ/°
82.718(3)
Volume/Å3
2802.4(2)
Z
4
r/g cm3
2.281
μ/mm–1
5.202
F(000)
1768.0
Crystal size/mm3
0.215×0.111×0.042
T/K
293(2)
Reflections collected
23270
Independent reflections
11443 [Rint = 0.0397, Rsigma = 0.0673]
Data/restraints/parameters
11443/0/559
GOF on F2
1.018
Final R indexes [I> = 2σ (I)]
R1 = 0.0545, wR2 = 0.1157
Final R indexes [all data]
R1 = 0.0975, wR2 = 0.1369
Max/Min eÅ–3
1.31/–1.39
Selected bond lengths (Å) and bond angles (°) for compound 1
Compound 1
Cd1–I1
2.7955(9)
Cd1–I2
2.7912(10)
Cd1–I3
2.7636(10)
Cd1–I4
2.6972(9)
Cd1’–I1’
2.8055(9)
Cd1’–I2’
2.7561(9)
Cd1’–I3’
2.7774(10)
Cd1’–I4’
2.7320(10)
I2–Cd1–I1
112.83(3)
I3–Cd1–I1
104.40(3)
I3–Cd1–I2
106.79(3)
I4–Cd1–I1
111.62(3)
I4–Cd1–I2
109.72(3)
I4–Cd1–I3
111.28(3)
I2’–Cd1’–I1’
109.47(3)
I2’–Cd1’–I3’
109.84(3)
I3’–Cd1’–I1’
107.33(3)
I4’–Cd ’–I1’
108.67(3)
I4’–Cd1’–I2’
110.77(3)
I4’–Cd1’–I3’
110.69(3)
(a) Schematic diagram of compound 1; (b) Hydrogen bond diagram of compound 1; (c) Stacked diagram of compound 1.
Results and discussion
Description of crystal structures
Crystal structure of compound [syn/syn–OM–IM–C] (CdI2) [syn/anti–OM–IM–C’] (Cd’I’2) (1)
It can be seen from single crystal diffraction and structural analysis that compound 1 belongs to the triclinic system and the space group is P–1. The smallest structural unit comprises: two parts of imidazole ring divalent cation (C22H22N4)2+ and (C’22H’22N’4)2+ and two parts of dianion (CdI4)2– and (Cd’I’4)2–. As shown in Fig. 1(a), At the room temperature, this cyclophane of compound 1 exist in two conformations anions, assigned as a flattened anti conformation in which the benzene rings may simply lie closer to the “plane” of the cyclophane and a syn conformation in which the imidazolium C2 protons are endo with respect to the cyclophane skeleton [9]. The two cationic conformations may be affected by temperature or anionic ligands. The two anionic moieties, Cd1 and Cd1’, are tetracoordinated and coordinate with four iodine atoms to form a tetrahedral configuration. Cd1–I1 = 2.7955(9)Å, Cd1–I2 = 2.7912(10)Å, Cd1–I3 = 2.7636(10)Å, Cd1–I4 = 2.6972(9)Å, Cd1’–I1’ = 2.8055(9)Å, Cd1’–I2’ = 2.7561(9)Å, Cd1’–I3’ = 2.7774(10)Å, Cd1–I4’ = 2.7320(10)Å, I–Cd1–I has a bond angle range from 104.40 (3)° to 112.83(3)°, I’–Cd1’–I’ has a bond angle range from 107.33(3)° to 109.84(3)°. C–H⋯I4’ = 2.9935Å (shown by the red dotted line in Fig. 1(b)), C–H⋯I3’ = 3.1031Å (shown by the blue dashed line in Fig. 1(b)). There is a hydrogen bonding interaction between imidazole macrocyclic cations and anions. They rely on these weak interaction forces to form a regular 2D supramolecular stacking structure, as shown in Fig. 1(c).
Powder X-ray diffraction (PXRD)
Powder X-ray diffraction has been used to confirm the purity of the samples in the solid state. For compound 1 the as-synthesized PXRD patterns closely match the simulated patterns generated from the results of the single crystal diffraction data, indicative of pure products which were shown in Fig. 2.
Power X-ray Diffraction of compound 1.
Theoretical calculation
We adopt Gauss View 6.0 program and B3LYP method [20–24] to investigate the electronic structure of the title compounds. A Lanl2dz basis set was used for Cd and I atoms. A 6–31G (d, q) basis set was applied in C, H, N atoms. The energy of compound 1 is –2327.72 au after one cycle of calculation. The dipole moment of title compounds is 69.40 Debye indicating a non-centrosymmetric structure. HOMO of energy crystal orbit in compound 1 is –2.7 eV and LUMO energy is –3.55 eV and –2.91 eV. The calculations based upon Density functional theory was carried out to obtain information about the HOMO and LUMO distributions of the compound 1. The molecular orbital amplitudes of compound 1 revealed that the HOMO is generally distributed on the (CdI4)2– anions, while the LUMO distributed mostly on the imidazole ring and benzene ring of the cation. The energy band gap calculated from the HOMO and LUMO energy values for compound 1 is 0.21 eV.∥
Thermogravimetric analyses
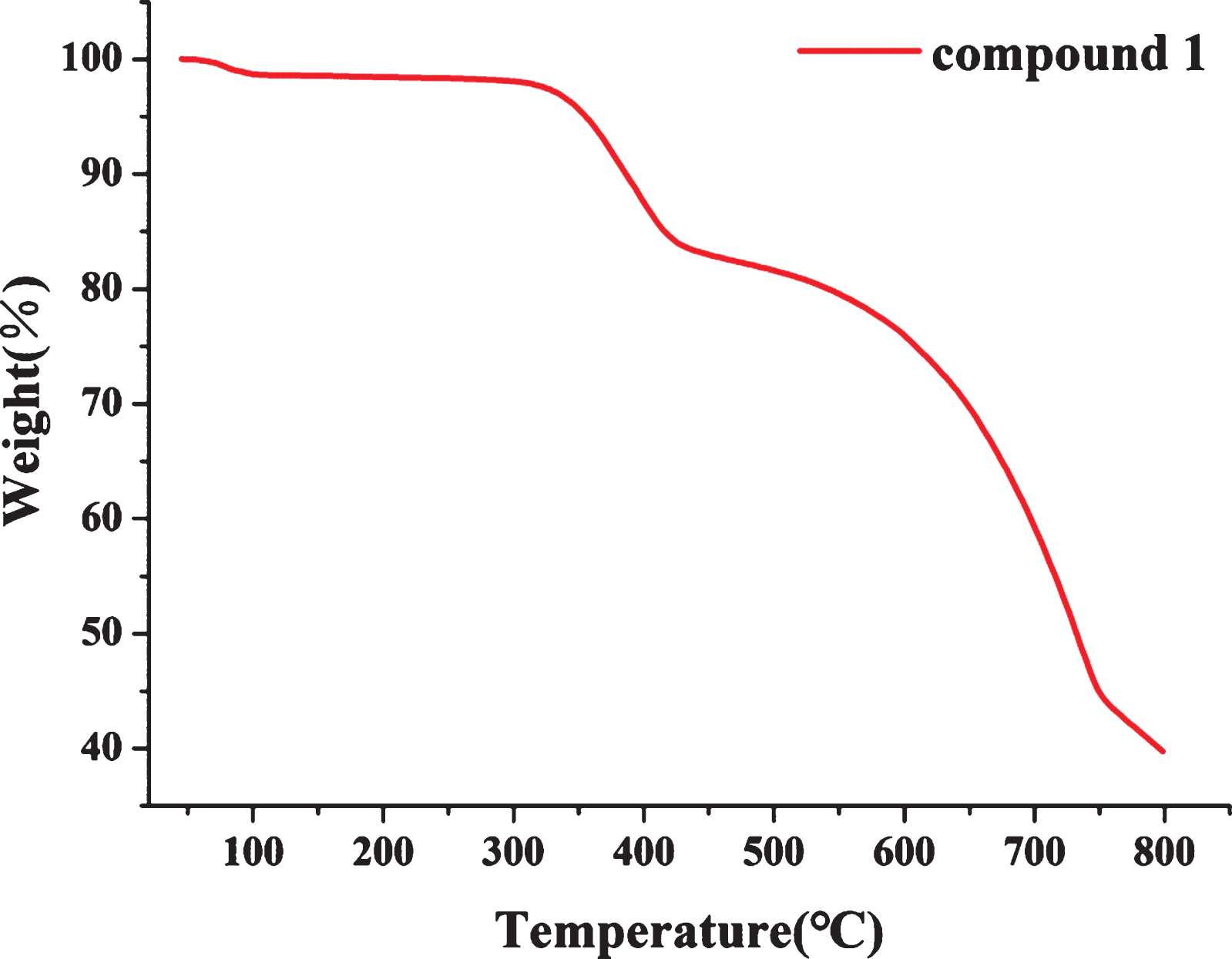
To investigate thermostability of compound 1, the thermogravimetric analyses (TGA) experiments were performed up to 800°C in a flowing N2 atmosphere, as shown in Fig. 3. Compound 1 is stable up to the temperature of around 350°C, indicating that it is a heat-resistant hybrid [19, 25]. The TG curves of compound 1, which is mainly undergoing two stages of weightlessness. Compound 1 has the largest mass loss at 350–390°C. The biggest reason may be the volatile decomposition of organic cations. The decomposition of compound 1 in the second part may be the decomposition of the cation causing the collapse of the anion structure. Therefore, the stability of compound 1 is relatively good. The stability of compound 1 may be due to the presence of hydrogen bonds interaction between the cationic ligands.
TG plot of compound 1.
Conclusion
In this article, we successfully synthesized one new mononuclear structure inorganic-organic hybrid supramolecule using imidazole macrocyclic divalent cation templated self-assembly. The conformation immobilization of cyclophane cation was realized through the metal anion recognition. Organic cation adapted to the anion by rotation of the imidazole ring and ring flip of the benzene rings. The conformation behavior of cyclophane in them was that the imidazole rings were mutually syn, whereas the benzene rings were anti and syn in compound 1. The stability of compound 1 may be due to the presence of hydrogen bond interaction.
Associated content
Supporting Information: Important crystallographic data (Tables 1 and 2), simulated powder XRD patterns (Fig. 2) for 1, comparison of important parameters of crystal structure of compound 1, CCDC reference numbers: 1893139 for 1. This data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: t441223336033 or E-mail: deposit@ccdc.cam.ac.uk.
Footnotes
Acknowledgments
Research efforts in the Niu’s group are supported by the National Science Foundation of China (No. 21671177).
References
1.
(a)
AhangarN.AyatiA.
and
AlipourE., Chem Biol Drug Des78(9) (2011), 844–852; (b)
GobisK.FoksH. and
BojanowskiK., Med Chem20(1) (2012), 137–144; (c)
BianchiniC.MantovaniG.
and
MeliA., Eur J Inorg Chem8 (2003), 1620–1631; (d)
LiF.W. and HouT.S., Adv Synth Catal350(14, 15) (2008), 2391–2400.
2.
(a)
MoorcraftL.P.MorandeiraA.
and
DurrantJ.R., Dalton Trans48 (2008), 6940–6947; (b)
SinghaD.K. and
MahataP., Inorg Chem54 (2015), 6373–6379; (c)
QiaoX.Y.LiZ.Y.LuY.B.XiaoM.YanZ.N.
and
NiuY.Y., Main Group Chem17 (2018), 133–146.
(a)
XiaoM.LuY.B.LiZ.Y.
and
NiuY.Y., J Cluster Sci29 (2018), 1039–1049; (b)
WangF.R.WangC.H.WuB.L.YanZ.N.NiuY.Y.
and
HouH.W., Main Group Chem. 17 (2018), 211–218. (c)
WangC.H.SongL.S.NiuY.Y. and
LiangY., Main Group Chem14 (2015), 71–78.