
Abstract
In this research, a DFT calculation was performed for study to investigate the encapsulation of the anticancer drug Ibrance into CNT(8,8-7) by using M062X/6-311G* level of theory in the solvent water. TD-DFT method was used to compute the electronic spectra of the Ibrance drug, CNT(8,8-7) and complex CNT(8,8-7)/Ibrance in aqueous medium for the study of non-bonded interaction effect. The non-bonded interaction effects of Ibrance drug with CNT(8,8-7) on the electronic properties and natural charges have been also studied. The results display the change in title parameters after process adsorption. According to NBO results, the molecule Ibrance and CNT(8,8-7) play as both electron donor and acceptor at the complex CNT(8,8-7)/Ibrance. Charge transfer, on the other hand, occurs between the bonding, antibonding, or nonbonding orbitals of Ibrance drug and CNT (8,8-7). According to QTAIM analysis and the LOL and ELF values, all intermolecular bonds in the complex are non-covalent in nature. As a result, CNT(8,8-7) can be thought of as a drug delivery system for transporting Ibrance as an anticancer drug within biological systems.
Introduction
Controlling the side effects and toxicity of drugs is an important and difficult task that is usually accomplished by administering chemical drugs to the target region of the body. Target drug delivery at the site of action can be accomplished through methods such as direct injection, catheter, gene gun, and so on. However, these systems demonstrate direct delivery, which is inconvenient for the patient and costly to perform in humans [1–4]. TDDS (target drug delivery systems) are currently being developed, which include chemical, physical, and biological modifications or functionalization with or without the use of carriers. Functionalization of drug delivery agents with carbon nanotubes (CNTs) has received a lot of consideration in research because of their exceptional physical properties. Functionalization of drug delivery agents is a safe and well-established method for improving the agents’ interactions with drugs [5–7].
Among the nanomaterials that have attracted interest for drug delivery, nanotubes are the most concerning nanovectors. CNTs are also chemically inert and oxidation and corrosion resistant. The safety of clinical application of CNTs as anti-tumor drug carriers has been a subject of concern in recent years. Due to the special nanostructural properties of CNTs, potential toxic effects and improved biocompatibility may be avoided to ensure clinical drug safety. To achieve these positive effects, it needs to clarify the mechanisms underlying CNT-induced toxicity to eliminate toxic nanometer formulations [8–10].
Nanoencapsulation of drugs increases their efficacy, specificity and targeting ability [11]. Nanocarriers protect their payload from premature degradation in the biological environment, enhance bioavailability, and prolong presence in blood and cellular uptake [12].
Ibrance (Fig. 1), also known as palbociclib, is a kinase inhibitor used to treat HR-positive and HER2-negative breast cancer. Cancer cells proliferate in an uncontrolled manner. It inhibits the cyclin-dependent kinases CDK4 and CDK6 selectively [13, 14]. Ibrance was the first CDK4/6 inhibitor to receive FDA approval as a cancer treatment [15].
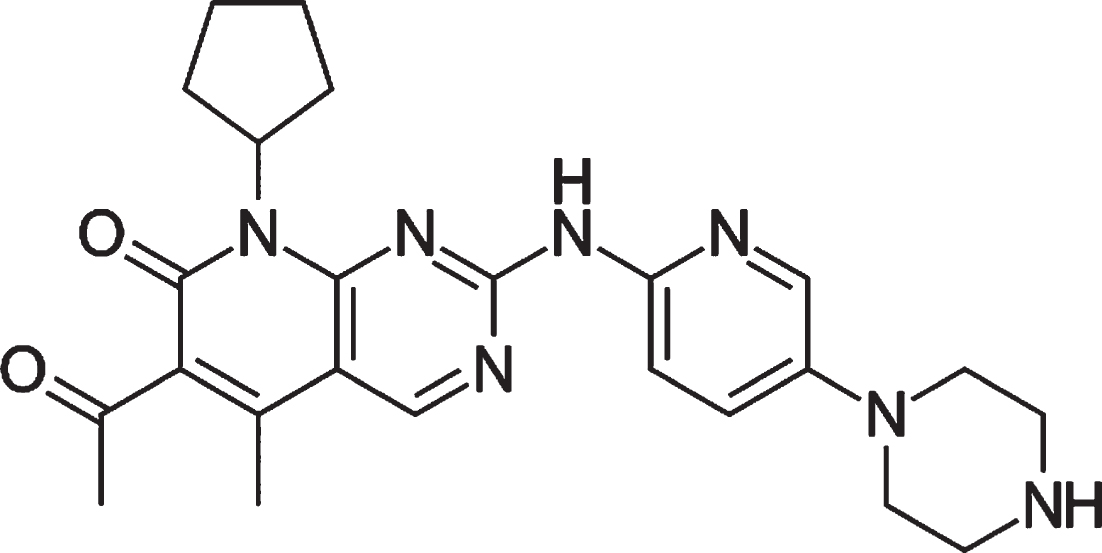
Chemical structure of Ibrance.
A thorough understanding of drug molecules’ interactions with carbon nanotubes is a necessary first step in the development of nanoscale drug delivery vehicles [16–18]. Many attempts have been made over the last decade to use density functional theory (DFT) calculations to analyze the interactions of CNTs with various biomolecules and drugs [19–21]. DFT studies may yield more useful information about the interaction of nanotubes [22–25].
In the present work, we used the DFT method to investigate the interaction between CNT and Ibrance drug. Frontier molecular orbitals, quantum-chemical molecular descriptors, Molecular Electrostatic Potential (MEP) analysis, natural charge, charge transfer analysis using Natural Bond Orbital (NBO) analysis, Quantum theory of atoms in molecules (QTAIM) and electronic structure and excited states were all investigated.
In current research, the encapsulation and non-bonded interaction of CNT(8,8-7) with the anticancer drug Ibrance in the solvent water was investigated. The solvent effect was calculated using the Polarized Continuum Model (PCM) [26]. The Gaussian 09W software [27] was used to perform quantum chemical calculations using DFT at the M062X/6-311G* level of theory for optimization of Ibrance drug, CNT(8,8-7) and complex CNT(8,8-7)/Ibrance. The choice of M062X functional is based on the fact that it produces acceptable results for adsorption energies [28]. The following equation was used to calculate the adsorption energy (Ead) [29]:
where ECNT (8,87)/Ibrance, ECNT (8,87) and EIbrance are energies of the CNT(8,8-7) with the adsorbed Ibrance, CNT(8,8-7) and Ibrance drug, respectively.
The molecular orbital (MO) calculations of the title systems such as EHOMO, ELUMO, energy gap between LUMO and HOMO (Eg) were also performed. GaussView 05 software [30] was used to visualize the optimized structures, HOMO, LUMO, and MEP surfaces. The quantum molecular descriptors for the Ibrance drug, CNT(8,8-7) and complex CNT(8,8-7)/Ibrance including ionization potential (I), electron affinity (A), global hardness (η), electronegativity (χ), electronic chemical potential (μ), electrophilicity (ω) and chemical softness (S) are computed by follows equations [31]:
In the following, the effects of encapsulation of Ibrance drug into CNT(8,8-7) on the natural charge [31] was studied. The electronic transitions of the molecule Ibrance and the complex CNT(8,8-7)/Ibrance were calculated using the Time-dependent Density Functional Theory (TD-DFT) method [31]. In order to understand hyperconjugative interactions and charge delocalization, the electronic structure of the title structures was also investigated using NBO analysis [32] at the M062X/6-311G* level. Electron delocalization from donor orbitals (full NBOs) to acceptor orbitals (empty NBOs) demonstrates a conjugative electron transfer process [31]. While electron transfer from donor orbital (i) to acceptor orbital (j) and delocalization process i→j, the stabilization energy E(2) is calculated [32]:
Quantum theory of atoms in molecules (QTAIM) analysis [33] was carried out by MultiWFN 3.1 [34].
Optimized structures
In the first step, we have optimized the compounds Ibrance, CNT(8,8-7) and complex CNT(8,8-7)/Ibrance in water using the M062X/6-311G* level of theory. The optimized structures are shown in Fig. 2.
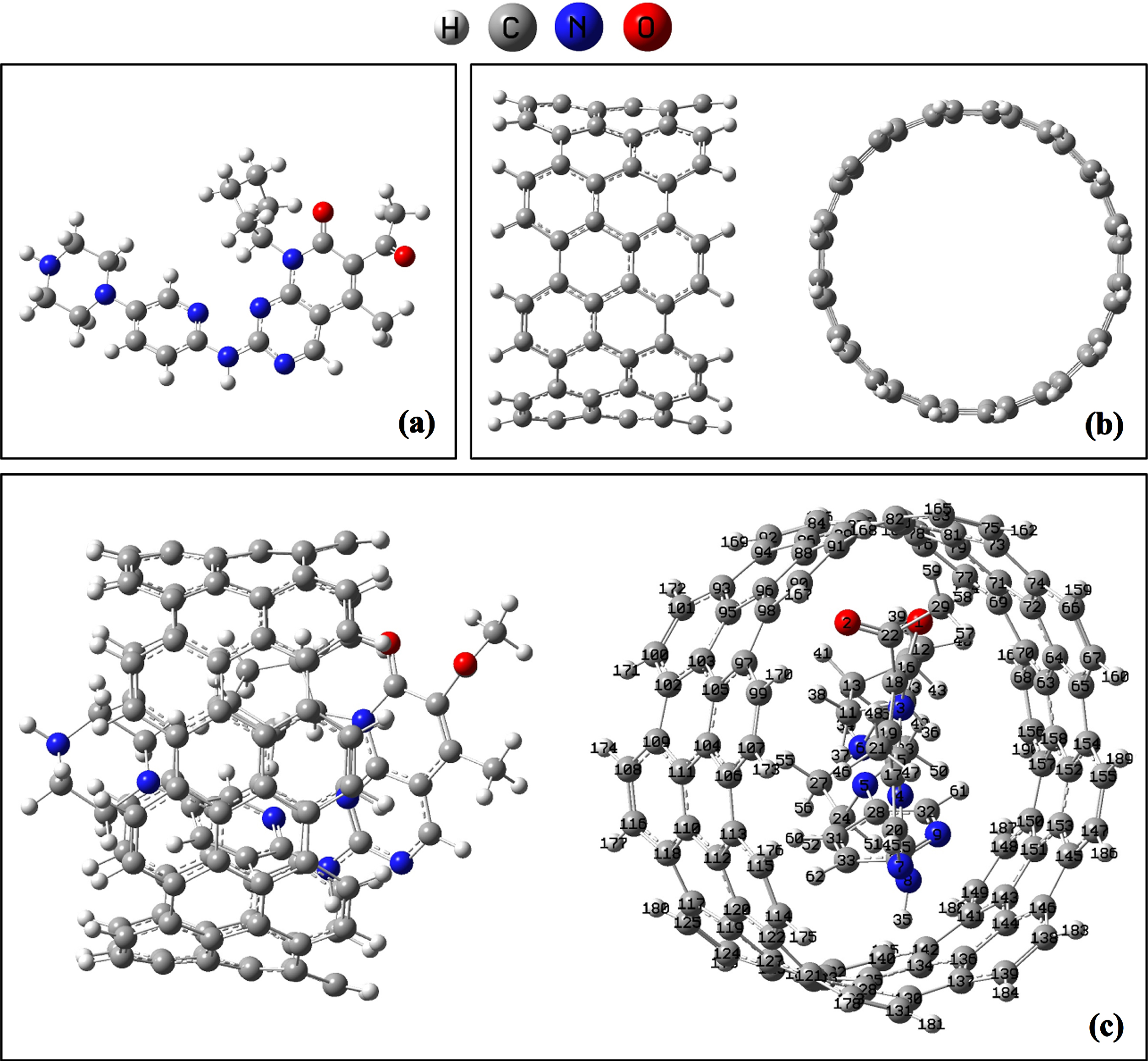
The optimized geometry of the molecules (a): Ibrance, (b): CNT(8,8-7) and (c): complex CNT(8,8-7)/Ibrance using M062X/6-311G* level of theory.
Table 1 shows the thermochemical parameters calculated for the molecules Ibrance, CNT(8,8-7) and complex CNT(8,8-7)/Ibrance with M062X/6-311G* level of theory. According to the reported results in Table 1, when Ibrance drug is encapsulated in CNT(8,8-7), the Thermal, Gibbs, and Enthalpy energies decrease. In the non-bonded intermolecular interaction with CNT, energy values indicate a decrease in reactivity and an increase in stability of Ibrance drug (8,8-7).
The thermochemical parameters of the molecules Ibrance, CNT(8,8-7), complex CNT(8,8-7)/Ibrance calculated by M062X/6-311G* level of theory
Geometrical parameters are critical in interpreting drug interactions with nanotubes in drug delivery systems. Table 2 summarizes the calculated bond lengths of Ibrance drug, CNT(8,8-7), and CNT(8,8-7)/Ibrance complex at the interaction sites. Some bond lengths of Ibrance drug and CNT are changed after interaction, according to the results. The bond lengths of N3-C10, N3-C16, N4-C15, N7-C25, N8-C30, C10-C12, C14-H44, C30-C33, C33-H62 in the molecule Ibrance is 1.480Å, 1.395Å, 1.329Å, 1.354Å, 1.480Å, 1.553Å, 1.091Å, 1.399Å, 1.083Å and after the interaction of Ibrance drug with CNT(8,8-7) changes to 1.487Å, 1.390Å, 1.314Å, 1.349Å, 1.436Å, 1.518Å, 1.086Å, 1.391Å, 1.076Å, respectively (see Table 2). These bonds show the high changes in Ibrance drug during interaction process with CNT(8,8,7). The optimized structure of CNT(8,8-7) has the C64-C72, C80-C81, C80-C82, C88-C89, C90-C98, C97-C105, C99-C107, C102-C109, C121-C122, C128-C129 bonds length 1.417Å, 1.417Å, 1.407Å, 1.407Å, 1.408Å, 1.407Å, 1.377Å, 1.461Å, 1.461Å, 1.417Å, while after interaction with Ibrance drug change to 1.421Å, 1.420Å, 1.409Å, 1.409Å, 1.405Å, 1.409Å, 1.379Å, 1.459Å, 1.465Å, 1.421Å, respectively. The geometry of the structures Ibrance drug and CNT(8,8-7) changes after the non-bonded interaction of the two compounds and the formation of the complex, according to the results, though these changes are not fundamental.
The calculated bond length (Å) of the molecule Ibrance, CNT(8,8-7) and complex CNT(8,8-7)/Ibrance using M062X/6-311G* method in a solvent water
NBO analysis is used to investigate intra- and intermolecular bonding as well as bond interaction in molecular structure [31]. The stabilization energy (E(2)) represents the amount of electron participation in the resonance between atoms of molecules [26]. The larger E(2) indicated that an electron donor has a high tendency to donate to an electron acceptor [26]. The NBO analysis for complex CNT(8,8-7)/Ibrance was performed using M062X/6-311G* level of theory and the results are shown in Table 3. According to obtained results, the π → π*, π → σ*, σ → π*, n→π*, n→σ*, π* → π*, π* → σ* transitions take place between Ibrance drug and CNT(8,8-7). The π*(N9-C30)→π*(C144-C146), π*(C17-C20)→π*(C106-C107) transitions have the highest resonance energies (E(2)) of 0.70 kcal/mol, 0.95 kcal/mol respectively, rather than other transitions Ibrance→CNT. The lone pairs (n) of the O1 and O2 atoms in the molecule Ibrance overlaps with the anti-bonding orbitals π* and σ* of CNT(8,8-7) that are including n1(O1)→π*(C75-C83), n1(O1)→π*(C89-C91) n1(O2)→σ*(C90-H167), n2(O2)→σ*(C99-H170) interactions with resonance energies (E(2)) of 0.33 kcal/mol, 0.48 kcal/mol, 0.32 kcal/mol, 0.44 kcal/mol respectively. The other important transitions of Ibrance→CNT are including π(O2-C22)→π*(C90-H167), σ(C14-H44)→π*(C78-C79), π*(N4-C15)→π*(C106-C107) interactions with stabilization energies (E(2)) about 0.49 kcal/mol, 0.34 kcal/mol, 0.44 kcal/mol, respectively. The maximum stabilization energy (E(2)) in charge transfer from CNT(8,8-7) to Ibrance is observed for The π(C130-C131)→σ*(N8-H35) and π*(C126-C127)→π*(C31-C33) transitions about 1.94 kcal/mol and 1.99 kcal/mol. The other important transitions of CNT→Ibrance are including π(C97-C105)→σ*(C11-H38), π(C120-C122)→σ*(C33-H62), π(C121-C123)→σ*(N8-H35), π(C128-C129)→π*(C31-C33), π(C144-C146)→π*(N9-C30) interactions with resonance energies of 0.39 kcal/mol, 0.83 kcal/mol, 0.48 kcal/mol, 0.96 kcal/mol, 0.49 kcal/mol, respectively. Thus, Ibrance and CNT(8,8-7) acts as both electron donor and electron acceptor; therefore, charge transfer takes place between they in the complex CNT(8,8-7)/Ibrance.
The donor-acceptor interactions and second-order perturbation energies (E(2), kcal/mol) related to charge transfer between Ibrance and CNT(8,8-7) in complex calculated using the M062X/6-311G* method
The donor-acceptor interactions and second-order perturbation energies (E(2), kcal/mol) related to charge transfer between Ibrance and CNT(8,8-7) in complex calculated using the M062X/6-311G* method
aE(2) Energy of hyperconjucative interactions. bEnergy difference between donor and acceptor i and j NBO orbitals. cF(i, j) Is the Fock matrix element between i and j NBO orbitals.
The frontier molecular orbitals (FMO), which include the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), play important roles in chemical reactions and charge transfer in molecular systems [29]. The HOMO and LUMO energies have the ability to donate and obtain electrons, respectively. The energy gap (Eg) between the HOMO and LUMO orbitals is a key parameter in determining electronic transport properties in molecular structures [24]. We have investigated the effects of non-bonded intermolecular interaction effects on the electronic properties of Ibrance drug and CNT(8,8-7). Table 4 displays the calculated results.
The calculated electronic properties of the Ibrance drug, CNT(8,8-7) and the complex CNT(8,8-7)/Ibrance at the M062X/6-311G* level of theory in the solvent water
The calculated electronic properties of the Ibrance drug, CNT(8,8-7) and the complex CNT(8,8-7)/Ibrance at the M062X/6-311G* level of theory in the solvent water
The adsorption energy (Ead) of the Ibrance drug over the CNT(8,8-7) is approximately 29.53 eV, indicating that the reaction is exothermic (Table 4).
The electron density in HOMO orbital of the compound Ibrance is primarily located on the double bonds (–C = C–and –C = N–) of pyridine ring, nitrogen atom of piperazine ring, double bonds (-C = N-) of imine group, whereas the LUMO orbital is localized on double the double bonds (-C = C- and –C = N-) of pyrimidine ring, carbonyl group (C = O) and double bonds (-C = N-) of imine group, as shown in Fig. 3. Thus, the contribution of pi (π) bonds and lone pairs accounts for the majority of charge transfer from the HOMO orbital to the LUMO orbital in the Ibrance drug. The HOMO and LUMO orbitals of the CNT(8,8-7) and complex CNT(8,8-7)/Ibrance are localized on the double bonds (-C = C-).
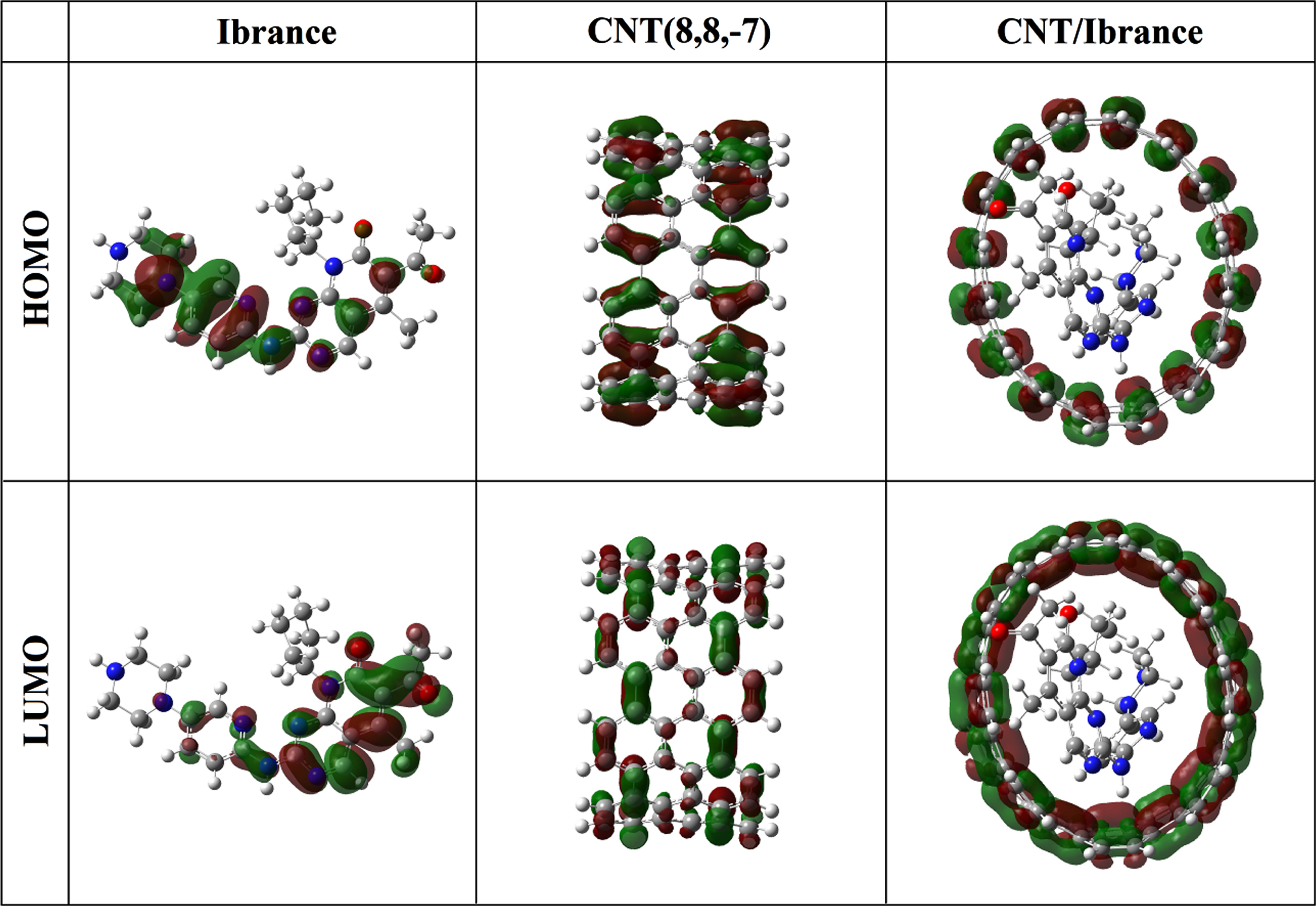
The calculated HOMO and LUMO orbitals of the compounds Ibrance, CNT(8,8-7), and complex CNT(8,8-7)/Ibrance at the M062X/6-311G* level of theory.
The energy gaps (Eg) of Ibrance drug is 5.68 eV, whereas after the encapsulation process of Ibrance drug into CNT(8,8-7) (complex) decreases to 3.29 eV. This result shows that the electrical conductivity of system complex CNT(8,8-7)/Ibrance is significantly higher than that of isolated Ibrance. The energy gaps of the investigated systems are also shown in the DOS plots [29] in Fig. 4.
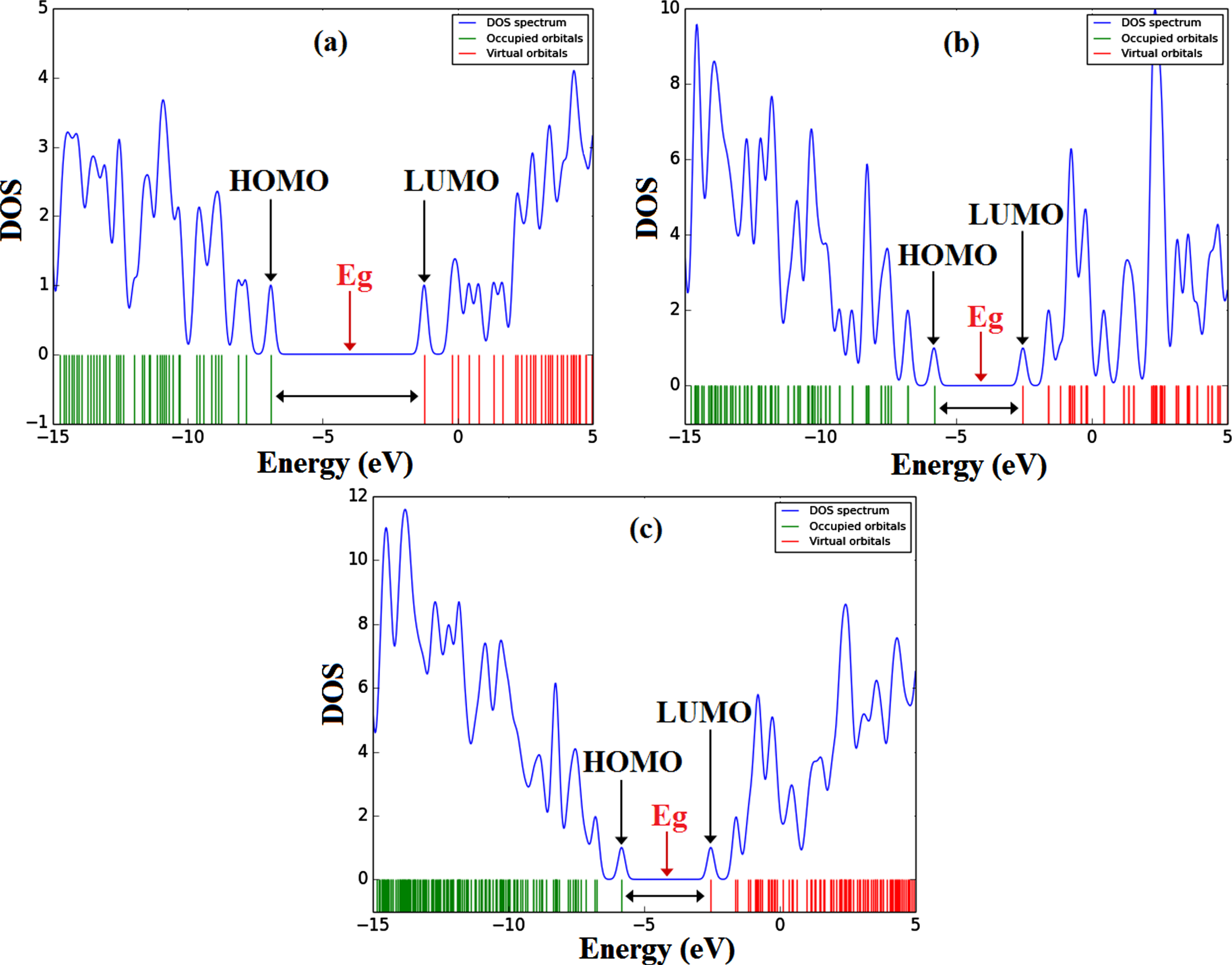
The DOS plot of the compounds Ibrance (a), CNT(8,8-7) (b), and complex CNT(8,8-7)/Ibrance (c) at the M062X/6-311G* level of theory.
The quantum molecular descriptors were reported in Table 4. The encapsulation of Ibrance into CNT changes its electronic properties (8,8-7). The hardness (η) of molecular structures is related to their stability, which is an important factor in detecting chemical reactivity [33]. The hardness and electronic chemical potential indexes of the complex CNT/Ibrance drug will be reduced, while electrophilicity and softness will be increased, as opposed to the isolated Ibrance drug. As a result, the complex CNT(8,8-7)/Ibrance exhibits high chemical activity, low chemical stability, and a soft molecular structure. As a result, the encapsulation of Ibrance drug into CNT(8,8-7) in solvent water alters electronic properties.
The values of dipole moment of CNT(8,8-7), Ibrance drug and complex CNT(8,8-7)/Ibrance are 0.031, 9.146, 5.629 Debye, respectively (Table 4). After capsulation process, the values of dipole moment of the CNT(8,8-7) decrease from 0.031 to 5.629 and in Ibrance drug increase from 9.146 to 5.629 Debye. The change of dipole moment after capsulation process show a charge transfer between Ibrance drug and CNT(8,8-7). The atomic charges have an important role on physical properties such as molecular polarizability, dipole moment, electronic structure and related properties of molecules [22]. Using the M062X/6-311G* level of theory, the NBO charges for equilibrium geometry of the CNT(8,8-7), Ibrance, and complex CNT(8,8-7)/Ibrance were calculated. Table 5 shows the calculated natural charges for selected atoms in three molecular structures (atoms are numbered according to Fig. 2). The natural charges of the O1, N4, N8, N9, C10, C11, C12, C13, C14, C37 atoms at the compound Ibrance are –0.674e, –0.584e, –0.606e, –0.483e, –0.013e, –0.395e, –0.392e, –0.383e, –0.383e, 0.205e, while after encapsulation process of Ibrance into CNT(8,8-7) change to –0.659e, –0.594e, –0.653e, –0.465e, –0.001e, –0.412e, –0.413e, –0.392e, –0.398e, 0.225e, respectively. The natural charges of the C68, C90, C97, C98, C102, C107, C109, C122, C123, C127, C128, C130, C131 atoms of CNT(8,8-7) are –0.176e, –0.176e, –0.030e, –0.030e, –0.030e, –0.176e, –0.030e, –0.030e, –0.176e, –0.013e, –0.013e, –0.030e, –0.176e respectively. After encapsulation of Ibrance into CNT(8,8-7), the natural charges change to –0.181e, –0.191e, –0.032e, –0.027e, –0.033e, –0.181e, –0.028e, –0.039e, –0.190e, –0.015e, –0.010e, –0.038e, –0.200e, respectively. Thus, it shows a charge transfer and non-bonded intermolecular interaction between Ibrance drug and CNT(8,8-7).
The natural charges (NBO charges, e) and NMR parameters (ppm) including CSI and CSA for the selected atoms in the molecules Ibrance, CNT(8,8-7) and the complex CNT(8,8-7)/Ibrance calculated by the M062X/6-311G* method
To investigate the interaction effect of Ibrance with CNT(8,8-7) on the λmax, we calculated the UV spectra of Ibrance drug and the complex CNT(8,8-7)/Ibrance in the solvent water using the TDM062X/6-311G* method, taking into account 20 excited states, and reporting the important transitions in Tables 6, 7. Tables 6, 7 display the λmax, oscillator strength (f), and excitation energies (E).
Electronic absorption spectrum of Ibrance drug calculated by TDM062X/6-311G* method
Electronic absorption spectrum of Ibrance drug calculated by TDM062X/6-311G* method
*In this Table the transitions with f≥0.10 are presented. *H: HOMO, L: LUMO.
Electronic absorption spectrum of complex CNT(8,8-7)/Ibrance calculated by TDM062X/6-311G* method
*In this Table the transitions with f≥0.10 are presented. *H: HOMO, L: LUMO.
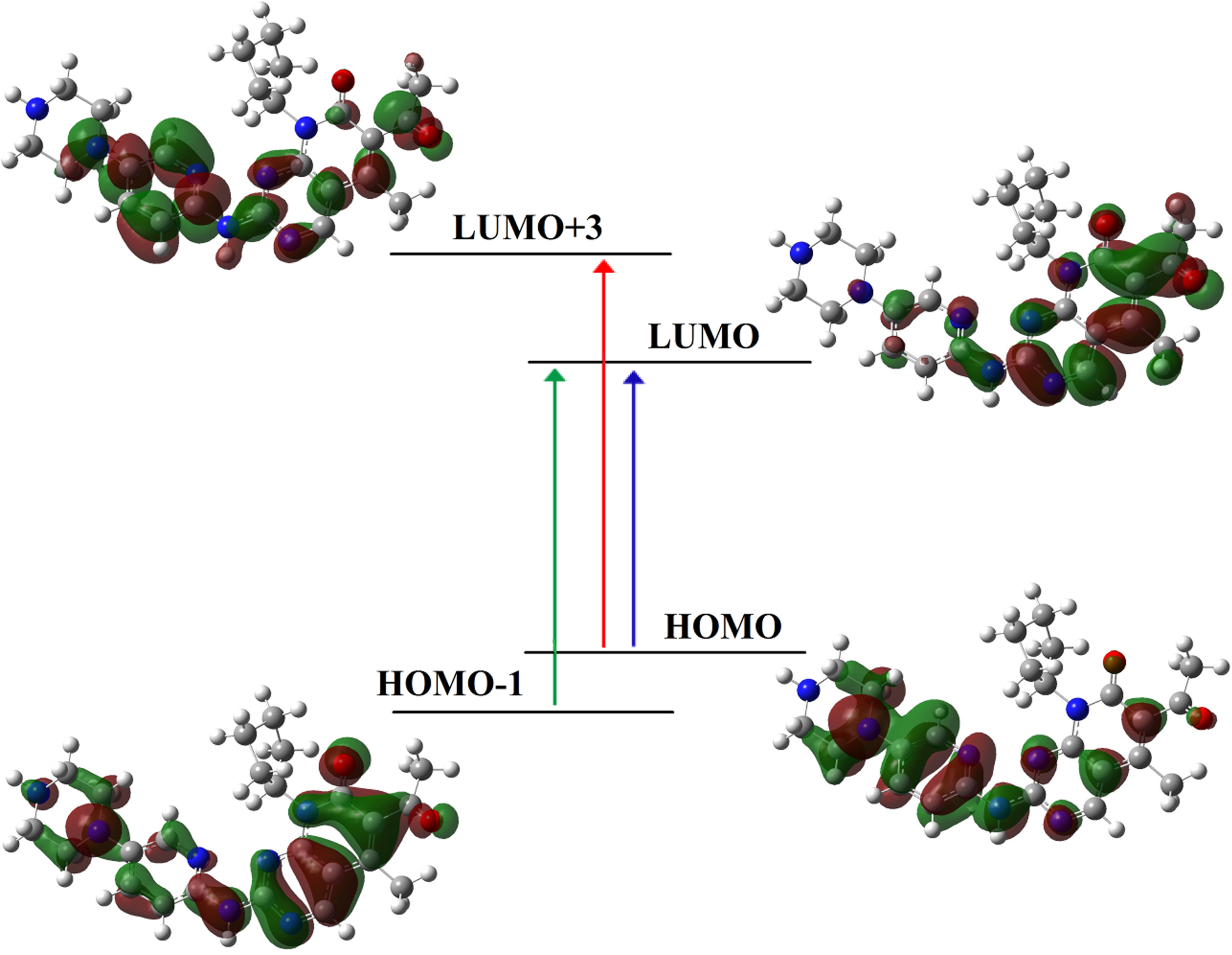
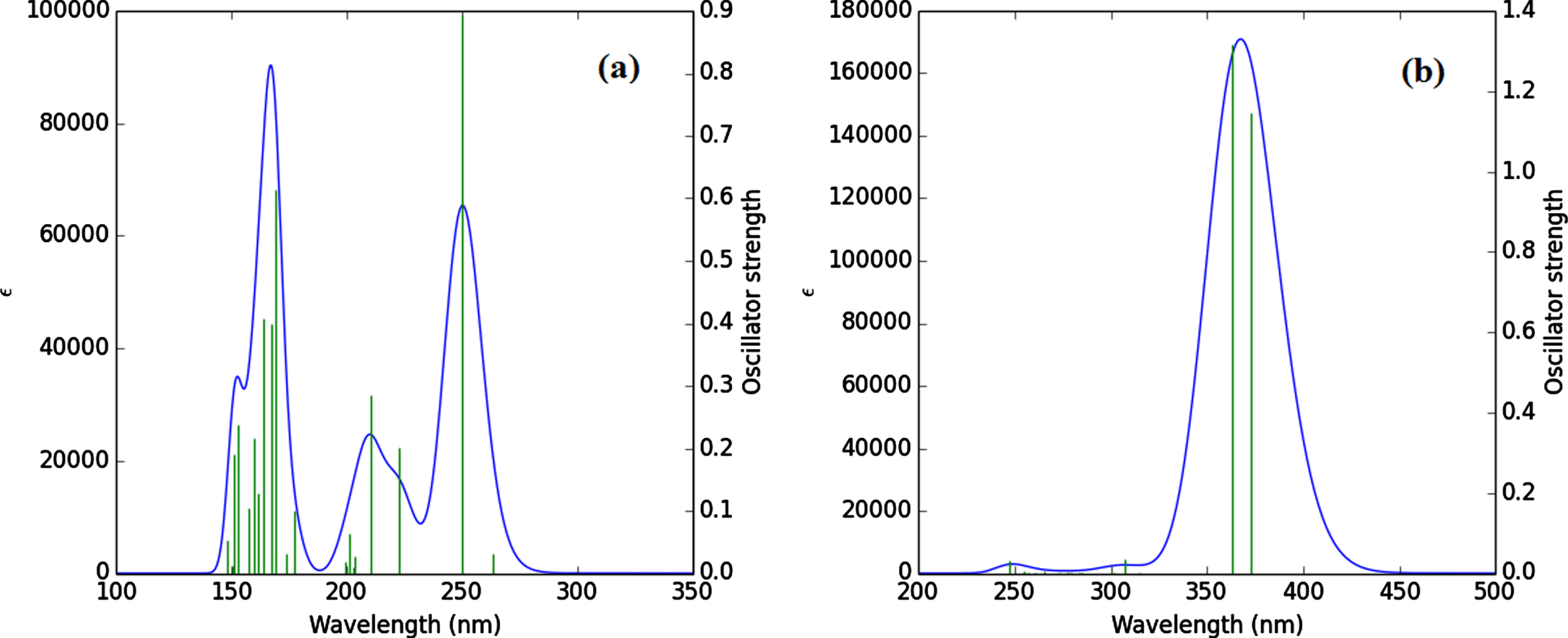
The calculated UV spectrum of Ibrance drug shows λmax at 250.05 nm (f = 0.89) (Table 6). The charge transfer at λmax = 250.05 nm takes place at excited state S0→S2 with three electron configurations including H-1→L (36%), H→L (45%), H→L + 3 (3%) in which the main transition is observed from HOMO to LUMO [H→L (45%)]. Figure 5 shows shape of molecular orbitals participants at λmax = 250.05 nm. As previously stated, the contribution of pi (π) bonds and lone pairs accounts for the majority of charge transfer from the HOMO orbital to the LUMO orbital in the Ibrance drug. The excited states of S0→S11, S0→S12, S0→S13 at 169.30 nm (f = 0.61), 167.7 (f = 0.39), 164.04 (f = 0.40) are also the other important excited state in the UV spectrum of Ibrance. The other excited states of Ibrance drug have low intensities and play no role in the formation of title compound electron spectrum (Table 6). Figure 6a depicts the theoretical electronic absorption spectrum of Ibrance drug in aqueous medium.
MO involved in formation of absorption spectrum of the complex Ibrance at λmax = 197 nm calculated by B3LYP/6-31G* method.
UV spectra of the Ibrance drug (a) and complex CNT(8,8-7)/Ibrance (b) calculated by TDM062X/6-311G* method.
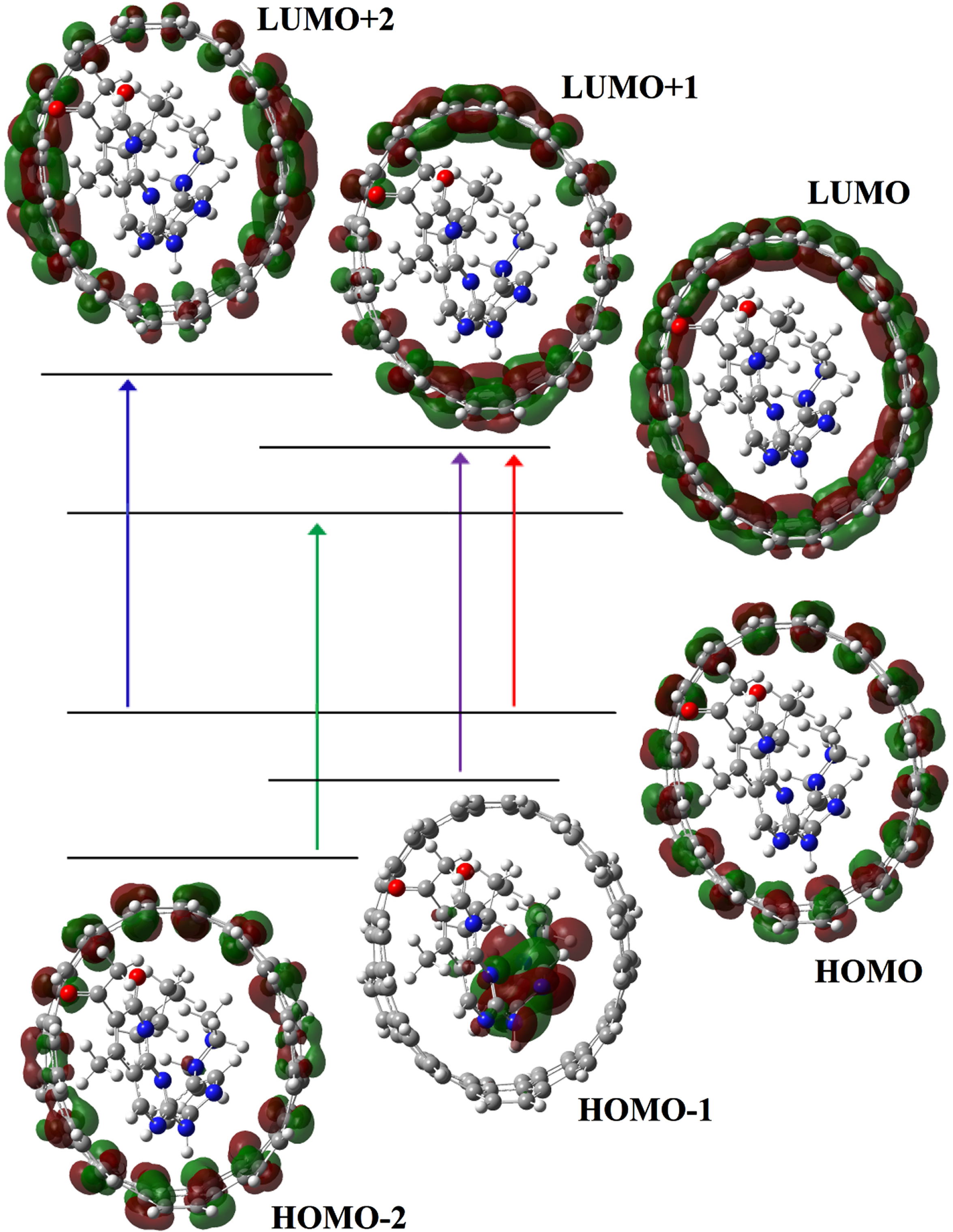
After the encapsulation of Ibrance drug into the CNT(8,8-7), λmax of complex CNT(8,8-7)/Ibrance appears at 373.02 nm (f = 1.14). The charge transfer at λmax = 373.02 takes place at excited state S0→S2 and is defined by four configurations such as H-1→L (37%), H→L + 1 (35%), H-2→L (2%), H→L + 2 (5%) (Table 7). The significant contribution to the absorption maxima is observed at HOMO-1→L transition [H-1→L], which contributes about 37% of the total excitations. Figure 7 shows shape of molecular orbitals participants at λmax = 373.02 nm. The excited state S0→S3 at 363.01 nm (f = 1.31) are also the other important excited state in the calculated UV spectrum of complex CNT(8,8-7)/Ibrance. The other excited states of the title compound have very small intensity (Table 7). Figure 6(b) shows the calculated UV spectrum of complex CNT/drug in the solvent water.
MO involved in formation of absorption spectrum of the complex Ibrance at λmax = 197 nm calculated by M062X/6-311G* method.
In the UV spectrum of the isolated Ibrance, λmax is observed at 250.05 nm, but after encapsulation in CNT(8,8-7) it is increased to 373.02 nm. As a result, we found that encapsulation and non-bonded intermolecular interaction of Ibrance into the CNT(8,8-7) change the value of λmax which can be interpreted as a bathochromic shift.
Quantum theory of atoms in molecules (QTAIM) [35] is used to reveal the nature of intermolecular interactions in the Ibrance/CNT complex at bond critical points (BCPs). Topological parameters such as electron density (ρ(r)), Laplacian of electron density (∇2ρ (r)), total energy density (H(r)), kinetic energy density (G(r)) and potential energy density (V(r)) [36] can be used to determine the intermolecular interactions. Figure 8 depicts the molecular graphs of the Ibrance drug molecule encapsulated in the CNT cavity. As shown in Fig. 8, there are five intermolecular hydrogen bonds formed between the hydrogen atom of the CNT and the oxygen atom of the Ibrance drug molecule (N4-H36, N5-H42O1-H58, O2-H167 and O2-H170). Topological analysis of the intermolecular bonds formed between Ibrance drug molecule and Carbon nanotube at the BCPs is listed in Table 8. The value of ρ(r) describes the strength of a chemical bond, so that a system with higher value of ρ(r) indicating more interaction and vice versa. The highest value of (r) is 0.099a.u, which is related to the C157-H50 bond. It demonstrates that this is the strongest bond. Positive and negative values of (r) characterize closed-shell and shared interactions, respectively [37, 38]. As shown in Table 1, the positive values of ∇2ρ (r) for all intermolecular bonds formed between Ibrance drug molecule and CNT indicate closed-shell interactions. The negative values of the ∇2ρ (r) and H(r) represent strong interactions, whereas the positive value of ∇2ρ (r) and the negative value of H(r) denote the medium interactions [39]. In the case of weak interactions, ∇2ρ (r) and H(r) values are positive. The calculated ∇2ρ (r) and H(r) values for all intermolecular bonds in the Ibrance drug/CNT complex are positive, showing weak interactions.
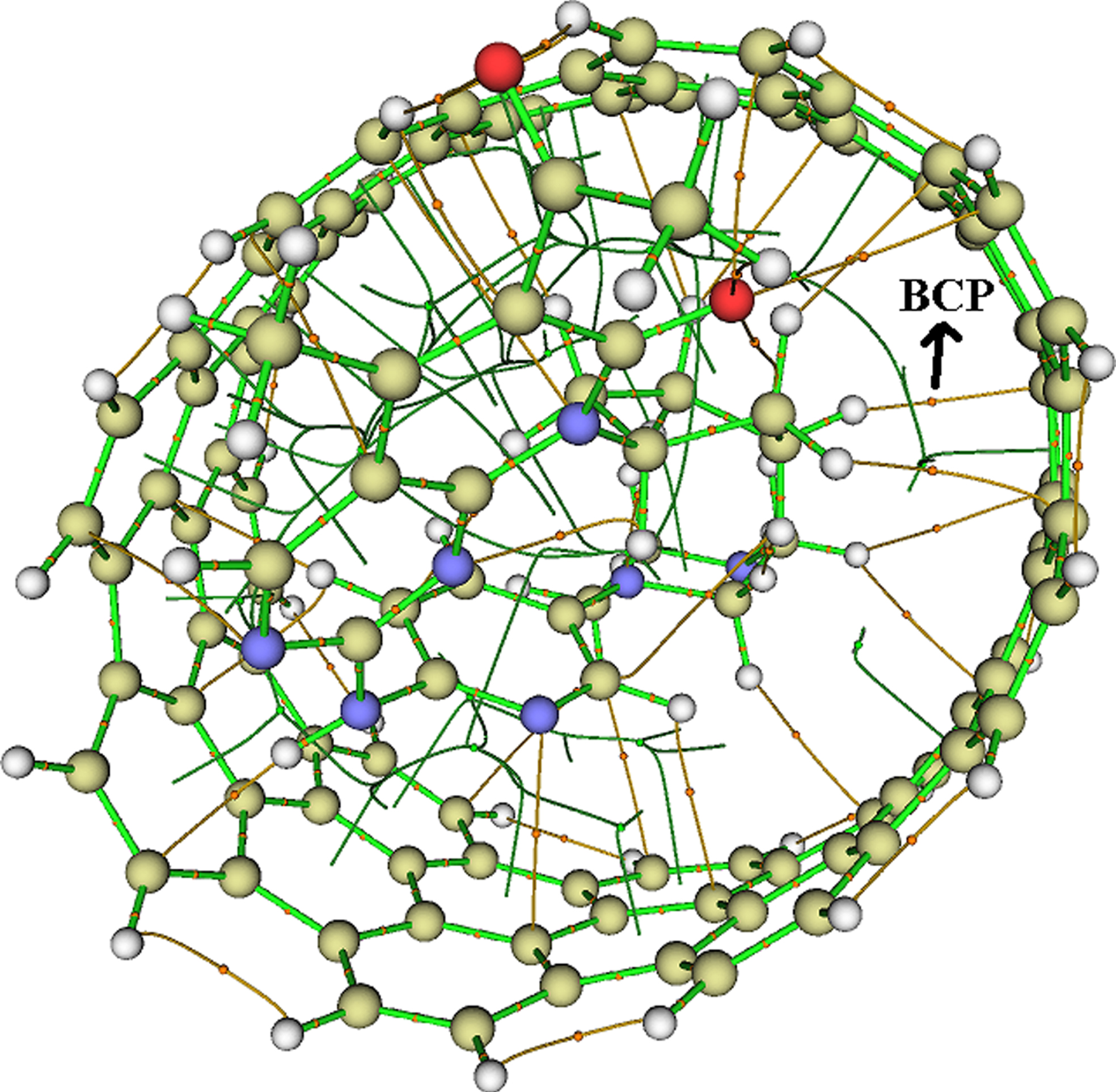
Molecular graphs of Ibrance/CNT complex.
Calculated topological parameters (in a.u) at the BCPs of the intermolecular bonds of Ibrance/CNT complex
The nature of the interactions can be determined based on the -G(r) and V(r) descriptors, so that the values of 0.5< -G(r)/V(r)<0, 1< -G(r)/V(r)<0.5 and -G(r)/V(r)>1 indicate covalent, electrostatic with partially covalent and non-covalent interactions, respectively [40]. As listed in Table 1, the -G(r)/V(r)value of all intermolecular bonds in the Ibrance drug/CNT complex is found to be greater than one, which means that they are non-covalent in nature.
Figure 9 depicts electron location function (ELF) and localized orbital locator (LOL) analyses of the studied complexes. The ELF and LOL are measures of the degree of electron localization. ELF and LOL values that are large (perfect localization, red areas) and small (low charge density, blue areas) indicate covalent and non-covalent interactions, respectively [41, 42]. The LOL and ELF values of all intermolecular bonds in the Ibrance/CNT complex are low, confirming non-covalent interactions between the Ibrance drug molecule and the CNT, as shown in Fig. 9.
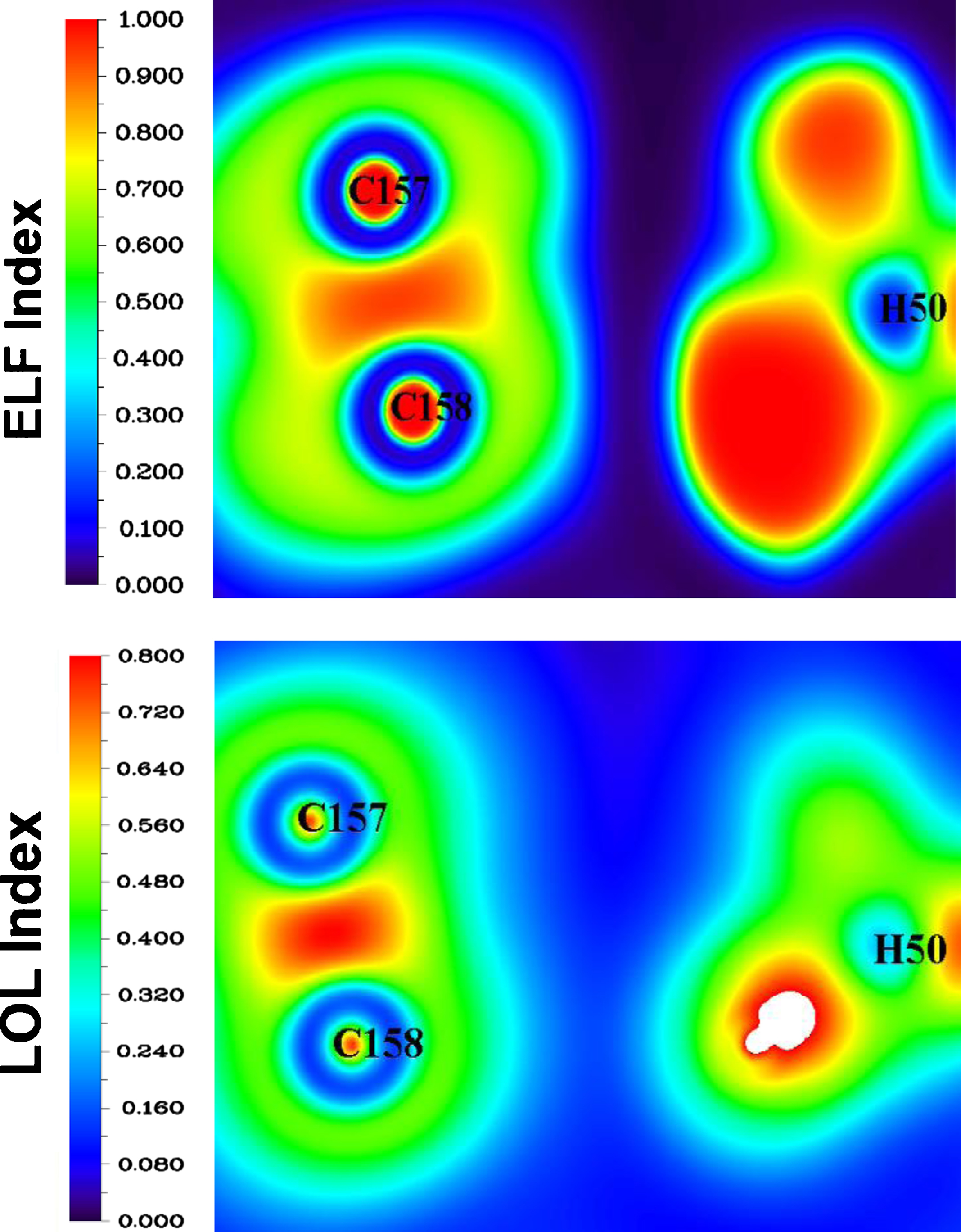
The ELF and LOL maps of molecular encapsulation of Ibrance drug molecule in CNT cavity.
Herein, the encapsulation of Ibrance drug into the CNT(8,8-7) was studied at the M062/6-311G* level of theory. According to the obtained results, the encapsulation process of Ibrance drug into the CNT(8,8-7) is an exothermic process and CNT(8,8-7)/Ibrance is a stable complex. It is found that some geometrical parameters of Ibrance and CNT(8,8-7) are changed after interaction between Ibrance and CNT. The NBO analysis predicted a charge transfer from the molecule Ibrance to CNT and vice versa. The energy gaps between LUMO and HOMO narrow after the interaction of Ibrance and CNT(8,8-7) rather than isolated Ibrance, and thus the electrical conductivity of the complex increases. After non-bonded interaction between Ibrance and the CNT(8), the electronic properties and natural charges change. The complex hardness and electronic chemical potential of the material will be reduced, while its electrophilicity and softness will be increased. The Non-bonded interaction between the compound Ibrance and CNT(8,8-7) changes the value of λmax. According to QTAIM analysis, the -G(r)/V(r) value of all intermolecular bonds in the Ibrance/CNT complex is found to be greater than one; therefore bonds are non-covalent. The LOL and ELF values of all intermolecular bonds in the Ibrance/CNT complex are low, confirming non-covalent interactions between the Ibrance molecule and the CNT. We hope that our findings can be can be used to improve drugs delivery to cancer cells using adsorbents.
Footnotes
Acknowledgments
The authors gratefully acknowledgment the financial and other support of this research, provided by the Islamic Azad University, Gachsaran Branch, Gachsaran, Iran.