
Abstract
Fetal lung interstitial tumor (FLIT) is a rare fetal malignancy that is typically diagnosed in the postnatal period, or, if recognized prenatally can mimic a benign lesion such as congenital pulmonary airway malformation. We present the earliest case of a FLIT tumor described by ultrasound and MRI at 26 weeks of gestation. Our case highlights features suggestive of FLIT including presentation later in gestation in combination with findings on fetal MRI such as a solid appearance with radiating curved bands of high signal within and along the periphery of the lesion (not as intensely high signal as the typical CPAM), possibly detailing a radiographic signature for these tumors. The role of betamethasone for these tumors is not known.
Keywords
Clinical case
A 37-year-old gravida 2 para 0102 female presented at 26 weeks of gestation for routine ultrasound (US) monitoring of a monochorionic-diamniotic twin gestation and was found to have a new echogenic mass in the left chest of Twin B (Fig. 1).
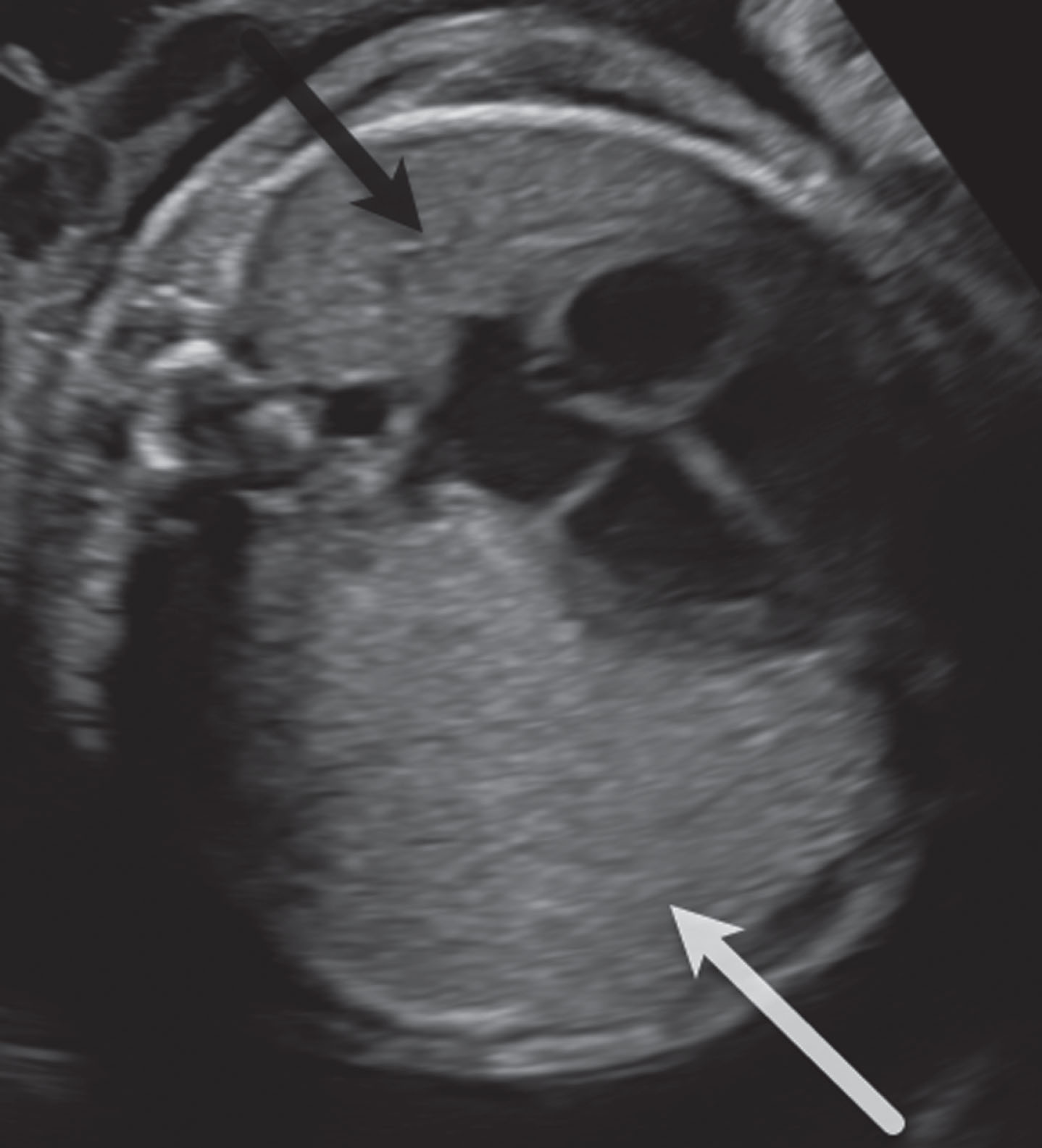
Transverse sonogram at 26 weeks gestation. The left lung mass (white arrow) is of uniform echotexture, only slightly more echogenic than the normal lung (black arrow) and has considerable mass effect, shifting the heart and mediastinum significantly to the right side. No cysts or aberrant feeding vessel were identified. There was no fetal hydrops or polyhydramnios.
The pregnancy resulted from in vitro fertilization secondary to maternal polycystic ovarian syndrome. First trimester serum screening was notable for an increased risk for Down Syndrome (1:142), however non-invasive prenatal screening was low-risk for aneuploidy. Surveillance for twin-twin transfusion syndrome, anatomic survey, and fetal echocardiography were unremarkable.
Given the interval development of the lung mass, the patient was referred to tertiary care center for additional fetal imaging. Fetal magnetic resonance imaging (MRI) and ultrasound (US) confirmed a 3.4×5.0×5.9 cm mass in the left upper lung with a CVR of 1.9 (CVR denotes CPAM volume ratio, or a ratio of the volume of the mass to gestational age). On US, the mass was characterized by minimally hyperechoic uniform echotexture and on T2W MRI, the mass had a slightly hyperintense appearance with curved and linear radiating bands of high signal within and along the borders. Contralateral mediastinal shift and flattening of the diaphragm were present (Figs. 1, 2a-b).
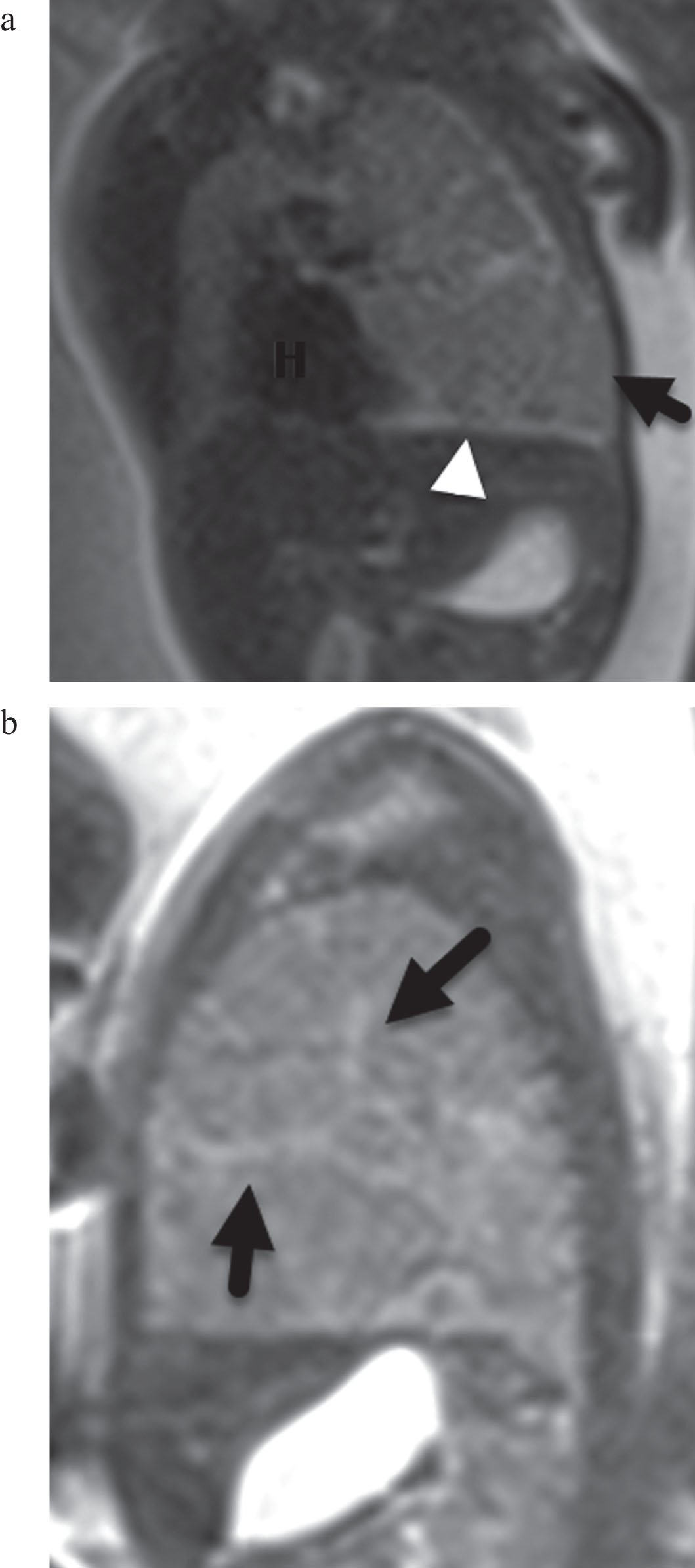
T2 haste MR images at 26 weeks gestation. (a) Coronal MR image. The left lung mass (black arrow) shows increased volume with mass effect shifting the heart (H) to the contralateral side and flattening the diaphragm. The overall signal is only slightly greater than the normal lung in contradistinction to the typical CPAM which is T2 hyperintense. There are linear zones of increased signal (white arrowhead) along the borders of the mass and within segments of the mass. (b) Sagittal MR image. The left lung mass signal is slightly greater than liver and muscle and contains multiple radiating bands of high T2 signal (black arrows). Note mass effect with flat diaphragm.
The differential diagnosis at that time included CPAM (congenital pulmonary airway malformation), nutmeg lung/lymphangiectasia or lobar hyperinflation. The patient received betamethasone at 27 weeks gestation and continued close monitoring throughout the pregnancy. The mass appeared stable in size without evidence of fetal hydrops or polyhydraminos. She received a second course of betamethasone at 33 weeks, with CVR in the third trimester decreasing to 0.8. At 36 weeks and 1 day of gestation, the patient underwent an uncomplicated repeat cesarean delivery in the setting of preterm labor. The affected neonate was noted to have significant respiratory distress at birth, with persistent requirement for non-invasive positive pressure ventilation and supplemental oxygen and was transferred to a tertiary care center for further evaluation and management.
Postnatal imaging, including frontal radiograph of the chest, demonstrated opacification of the left lung (Fig. 3a). Computed tomography (CT) scan of the chest demonstrated a large, primarily solid mass in the left upper lobe measuring 3.0×4.8×6.0 cm with mediastinal shift (Fig. 3b). Given the persistence of clinical symptoms, the neonate underwent left thoracotomy and left upper lobectomy on day of life 20. Post-operatively, respiratory support was quickly weaned, with resolution of symptoms, and the infant was discharged home on room air. The neonate remains healthy and asymptomatic at one-month follow-up.
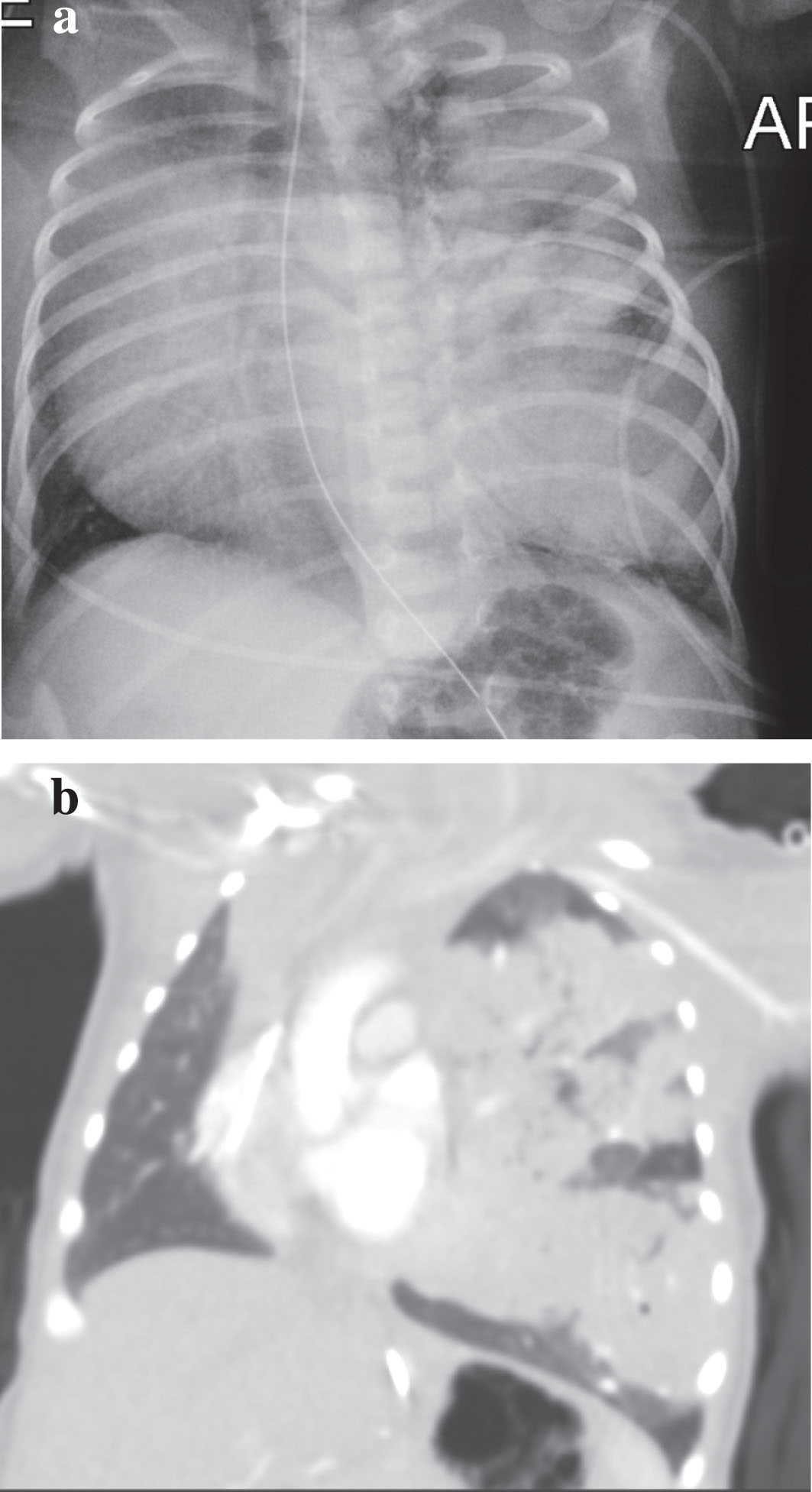
Day of life 1: postnatal imaging denoting globular mass in the left upper lobe. (a) Frontal Chest X-Ray. (b) Coronal CT.
Surgical pathology of the resected lung revealed a spongiform, hamartoma-like lesion composed of variably-sized cystic/expanded air spaces with diminished branching and increased interstitial thickness. The interstitium contained uniformly bland mesenchymal cells with ovoid nuclei, clear cytoplasm, and well-defined cytoplasmic borders (Fig. 4a-b).The cytoplasm of the interstitial cells also demonstrated diastase-digestible, cytoplasmic periodic acid-Schiff (PAS) positivity, indicating the presence of cytoplasmic glycogen (Fig. 4c-d). These microscopic features are consistent with fetal lung interstitial tumor (FLIT) [4].
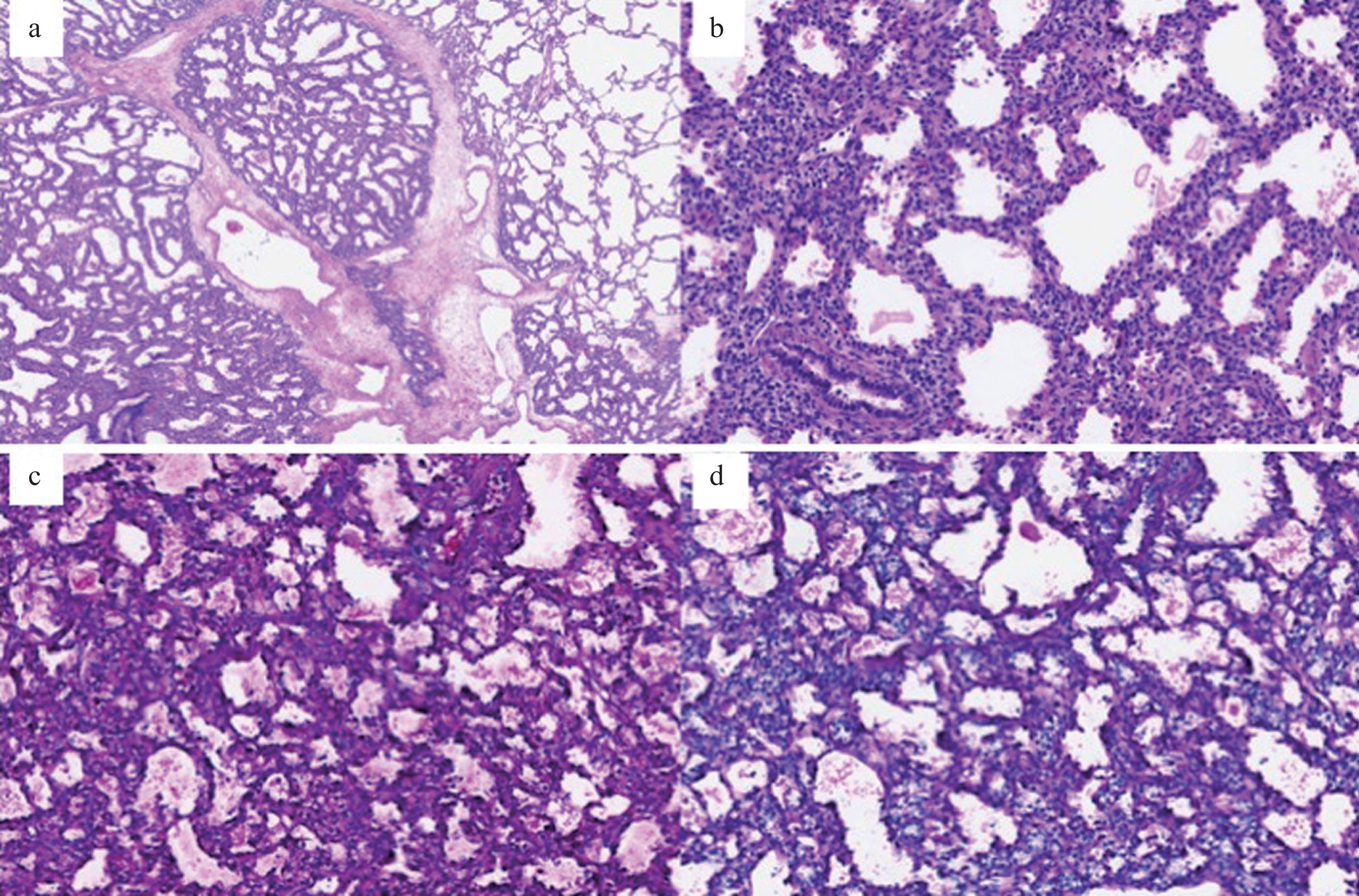
Histologic sections of fetal lung. (a) Lesional lung with expanded airspaces and thickened interstitium interfaces with normal alveolar spaces in the upper right corner (Hematoxylin and eosin). (b) The interstitium is expanded by uniformly bland mesenchymal cells with ovoid nuclei, clear cytoplasm, and well-defined cytoplasmic borders. Proteinaceous and cellular debris is present in some cystic spaces. (c) PAS positivity in the cytoplasm of the interstitial cells. (d) Diastase digestion removes the cytoplasmic PAS positivity which indicates the presence of cytoplasmic glycogen.
The differential diagnosis of prenatal thoracic lung lesions is broad and includes both benign congenital malformations such as CPAM types 1–3, pulmonary sequestration, bronchial atresia or bronchogenic cysts and malignant fetal tumors such as pleuropulmonary blastoma (PPB), congenital peribronchial myofibro-blastic tumor (PBFT) and FLIT [1, 2]. The most commonly observed thoracic lung lesion is the CPAM, estimated at 1 per 8,000 to 35,000 live births [1, 2]. Less commonly, fetal lung lesions may also result from pulmonary sequestration, bronchial atresia, or congenital lobar hyperinflation. While the majority of lung lesions are benign, malignant tumors have also been described, but are exceedingly rare; primarily PPB (less than 500 known cases reported), as well as PBFT [3]. FLIT, first described by Dishop, et al. in 2010, is the rarest fetal malignancy, with only few reported cases in the literature, of which only 3 were first identified prenatally. It is characterized by immature interstitial cells that resemble fetal lung tissue at 20–24 weeks of gestation. With resection of the tumor, no cases of recurrent disease have been observed to date [4].
Many fetal lung lesions can be readily identified via standard prenatal imaging. CPAM is characterized by unilobar mass involvement with a homogeneous hyperechoic appearance on obstetric US in the microcystic type or heterogeneous with visible variable sized cysts in the macrocystic type. With fetal MRI, the macrocystic CPAMs are generally characterized as T2 hyperintense (relative to the surrounding healthy lung tissue) with distinct cystic structures, while the microcystic CPAM is described as being heterogeneously hyperintense [1]. FLIT is not well characterized on pre- or post-natal imaging and can appear nearly identical to microcystic CPAM, particularly type 3 [5, 6]. To date, only three antenatal cases of FLIT have been reported all of which were retrospectively identified after post-natal pathologic confirmation and none have been identified early in the second trimester [1, 2]. The prenatal diagnosis of FLIT remains limited due to both the rarity of disease and predominant presentation in the postnatal period [5].
Classically, benign congenital malformations of the lung develop early in gestation, such as microcystic CPAMs that grow most rapidly during the canalicular phase of development (16–26 weeks of gestation) or such as bronchopulmonary sequestration thought to originate earlier in gestation during the pseudoglandular stage (prior to 17 weeks gestation) [1 , 7]. Thus, these lesions are frequently visualized on routine mid-second trimester screening. This is distinct from malignant congenital lung lesions or tumors, thought to develop later in gestation [5]. Furthermore, the developmental trajectory of benign lung lesions suggests that the majority of lesions regress in size prior to delivery [8, 9].
Recent evidence suggests that there may be a genetic basis to the development of FLIT. Anaplastic lymphoma kinase (ALK) is an enzyme in the tyrosine kinase family, and mutations in the ALK gene have been associated in a number of pediatric tumors, including inflammatory myofibroblastic tumors (IMT) and non-small-cell lung tumors. IMT, a rare tumor that most commonly affects the respiratory system, is characterized by both atypical and reactive cells with significant inflammatory findings. Indeed, molecular analysis of FLIT tumors has identified novel ALK mutations, namely a novel ALK-A2M gene fusion (α-2-macroglobulin). This novel ALK-A2M fusion suggests that some infantile pulmonary lesions may represent a sub- type of IMT that develops exclusively in neonates and infants [10, 11].
The antenatal administration of maternal steroids for certain lung lesions, particularly microcystic CPAMs, has been associated with improved outcomes. Associated effects include a plateau in growth of the lesion with an observed decrease in CVR, resolution of hydrops, and improved survival [12 –14]. Maternal steroids have also been administered in pregnancies with non-CPAM congenital lung lesions, namely bronchopulmonary sequestration, congenital lobar emphysema and bronchial atresia, however, given the small sample size, the potential impact on the natural history of these lesions remains unclear [12]. No current data exist describing response or outcomes in patients who received antenatal steroids in the setting of fetal lung malignancies and there is no current recommendation for or against administration in these cases.
To our knowledge, this is the earliest case of FLIT detected at 26 weeks of gestation characterized with US and fetal MRI and the first case of FLIT presenting in a twin gestation.
Conclusion
FLIT is a rare primary lung tumor that on prenatal imaging mimics CPAM type 3. Features suggestive of FLIT include presentation later in gestation in combination with findings on fetal MRI such as a solid appearance and mass effect with radiating curved bands of high signal within and along the periphery of the lesion (not as intensely high signal as the typical CPAM), possibly detailing a radiographic signature for these tumors.
Disclosure Statement
The authors report no conflict of interest and no financial disclosures.