
Abstract
Joubert syndrome is a rare neurological manifestation usually present in late infancy or early childhood with characteristic episodes of abnormal breathing pattern along with the neurological and other systemic involvement.We report a case of confirmed Joubert syndrome present in the immediate neonatal period with isolated spells of oxygen desaturations not accompanied by the classically described breathing pattern and absent neurological symptoms causing delay in the diagnosis. Isolated oxygen desaturation episodes could be a presenting manifestation of Joubert syndrome in a neonatal period.
Introduction
Joubert syndrome (JS) is a well-described neurological manifestation characterized by abnormal breathing pattern of episodic hyperpnoea followed with an apneic interval [1]. Other classical signs are hypotonia, motor involvement, abnormal ocular and tongue movements, and coordination problems [1]. On magnetic resonance imaging brain, a classic molar tooth appearance of mid brain is the pathognomonic sign [1, 2]. We present a newborn presenting initially with an isolated episodes of oxygen desaturations both at rest, and with oral feeds and with no other classical JS signs delaying the diagnosis. This case describe a variation of the clinical respiratory pattern in a newborn with JS.
Case presentation
A 3115 grams Hispanic male infant was born at 39 3/7 weeks of gestation to a 41 year old gravida (G) 5 para (P) 4 mother by an uncomplicated spontaneous vaginal delivery. The Apgar score was 8 and 8 at 1 and 5 minutes respectively. Rupture of membrane was around 17 hours and the amniotic fluid was clear. Group B streptococcus (GBS)screen was positive but mother received adequate penicillin prophylaxis. Prenatal course was also uncomplicated with normal prenatal anatomical scan. Mother denied use of medications during pregnancy, no history of drug or alcohol use. She has three other kids with the same partner and all kids are healthy. No family history of any genetic disorder. Quad screen done for advance maternal age was also negative. Initial physical examination was normal except for right pre-auricular pit and left hand postaxial polydactyly. Post-delivery, baby stayed with mother for routine care.
Around nine hours of life, baby was noted to have retractions and grunting and was transferred over to the neonatal intensive care unit for evaluation and management. Chest x-ray and blood gas were normal. Provisional diagnoses of possible sepsis vs. transient tachypnea of newborn ware made and baby was provided respiratory support with nasal CPAP. Baby was started on an empirical antibiotics (ampicillin and gentamicin) after getting blood culture. Baby subsequently improved with resolution of symptoms, and was successfully weaned to room air the following day. On day three of life, baby was noted to have intermittent desaturations along with perioral cyanosis and bradycardia. Most of these episodes were self-resolving but some required blow by oxygen and stimulation. Antibiotics were continued for seven days for possible clinical sepsis although the initial blood culture was negative and screening CBC and CRP were also normal. Cardiac work up was done for these cyanotic spells. EKG was within normal limits. Echocardiogram revealed mild hypoplastic aortic arch, bicuspid aortic valve, mild peripheral pulmonic stenosis, and trace of tricuspid valve regurgitation which however doesn’t explain these cyanotic spells. Blood gases were also negative for any respiratory or metabolic acidosis. Head ultrasound that was done to investigate for any CNS abnormality and bleeding, was also negative. During the course of his next few days, baby continued to have these random desaturations events, some during rest and some during feeds. Upper airway evaluation was negative for any abnormality. Barium swallow test was done to rule out reflux and was also negative. Later on baby was noted to have mildly reduced overall tone with droopy eyelids leading to underlying neurological pathology. MRI of brain that was done to rule out any central and brainstem pathology showed hypoplastic cerebellar vermis, molar tooth shaped midbrain, and atrophy of the right hippocampal gyrus suggestive of possible JS. Genetic evaluation was done thereafter to confirm the diagnosis. The infant karyotype was normal 46XY, microarray was normal, and JS panel was positive for homozygous mutation in RPGR1PIL gene confirming the diagnosis of JS.
Discussion
JS is a very rare autosomal recessive condition with an estimated incidence between 1 in 100,000 live births [2] and common in Ashkenazi Jewish, French-Canadians, and Hutterite ethnic populations [3 –5]. It was first reported by Marie Joubert in 1969 in a 6-month-old infant with an abnormal, rapid breathing pattern and developmental delay [6]. It is usually diagnosed in the infancy or the early childhood period and affects both males and female equally. Until date more than 200 cases have been reported with only two cases identified in prenatal period [7]. In the immediate newborn period, affected infants can present with an abnormal breathing pattern, characterized as episodic hyperpnoea followed with an apneic interval. Respiratory rates of up to 230 per minutes have been reported followed with apnea lasting up to one minute, mostly without any associated bradycardia or cyanosis. Other classical signs like hypotonia, motor involvement, abnormal ocular and tongue movements, and coordination problems usually present late and leads to delayed diagnosis [8, 9].
Our immediate provisional diagnoses in this case were possible sepsis including pneumonia, cyanotic cardiac lesions, reflux, intracranial bleeds, and upper airway abnormality. Work up for all these possible diagnoses was negative. Brain ultrasound was also reported normal.
In this case, the infant was fine the first day except the respiratory symptoms which resolved quickly with intervention. He didn’t exhibit the described classical breathing pattern of hyper apnea followed with apnea. He had these random episodes of desaturations up to 70’s, mostly with feeds and some at rest. He didn’t exhibit the signs of feeding in coordination except some inconsistent episodes of desaturations while feeding, and was able to finish off his feeds orally in a desired time. However, during his NICU course, hypotonia became more apparent along with ptosis triggering the possible underlying neurological pathology as a possible cause of his random desaturation events.
Classic JS is characterized by three primary findings: A distinctive cerebellar and brain stem malformation called molar tooth sign (MTS), hypotonia, and developmental delays [6, 9]. Often these findings are accompanied by episodic tachypnea or apnea and/or atypical eye movements (ocular motor apraxia). In general the breathing movements improve with age, truncal ataxia develop over time and acquisition of gross motor movements is delayed. Cognitive abilities are variable, ranging from severe intellectual disability to normal. Additional findings can include retinal dystrophy, renal disease, ocular colobomas, occipital encephalocele, hepatic fibrosis, polydactyly, oral hamartomas and endocrine abnormalities [9 –11].
Distinctive facial features can also occur in JS, these include a broad forehead, arched eyebrows, ptosis, epicanthial folds, anteverted nares, wide spaced eyes (hypertelorism), low set ears and triangle shaped mouth. The hallmark feature of JS is a combination of brain abnormalities that together are known as MTS setected on magnetic resonance imaging (MRI). The sign results from the abnormal development of structures near the back of the brain, including the cerebellar vermis and the brain stem. Brain MRI without contrast is the golden standard test for diagnosis [10 –12]. Mode of inheritance is predominantly autosomal recessive with few cases inherited as x-linked disorder. To date, more than 30 genes(INPP5E, TMEM216, AHI1, NPHP1, CEP290 (NPHP6), TMEM67 (MKS3), RPGRIP1L, ARL13B, CC2D2A, OFD1, TTC21B, KIF7, TCTN1, TCTN2, TMEM237, CEP41, TMEM138, C5orf42, TCTN3, ZNF423, TMEM231, CSPP1, and PDE6D) have been shown to cause JS related disorders (JSRD’s) leading to different phenotypic variations (Table 1) [13, 14]. The proteins produced from these genes affect cell structures called primary cilia. Primary cilia are microscopic, finger-like projections from the surface of cells and are involved in sensing the physical environment and in chemical signaling. Primary cilia are important for the structure and function of many types of cells, including neurons and certain cells in the kidneys and liver. Primary cilia are also necessary for the perception of sensory input, which is interpreted by the brain for sight, hearing, and smell [15].
Characteristic of different variation of Joubert syndrome along with common genes involved. JS – Joubert syndrome. Adapted from Brancati et al [14]
Characteristic of different variation of Joubert syndrome along with common genes involved. JS – Joubert syndrome. Adapted from Brancati et al [14]
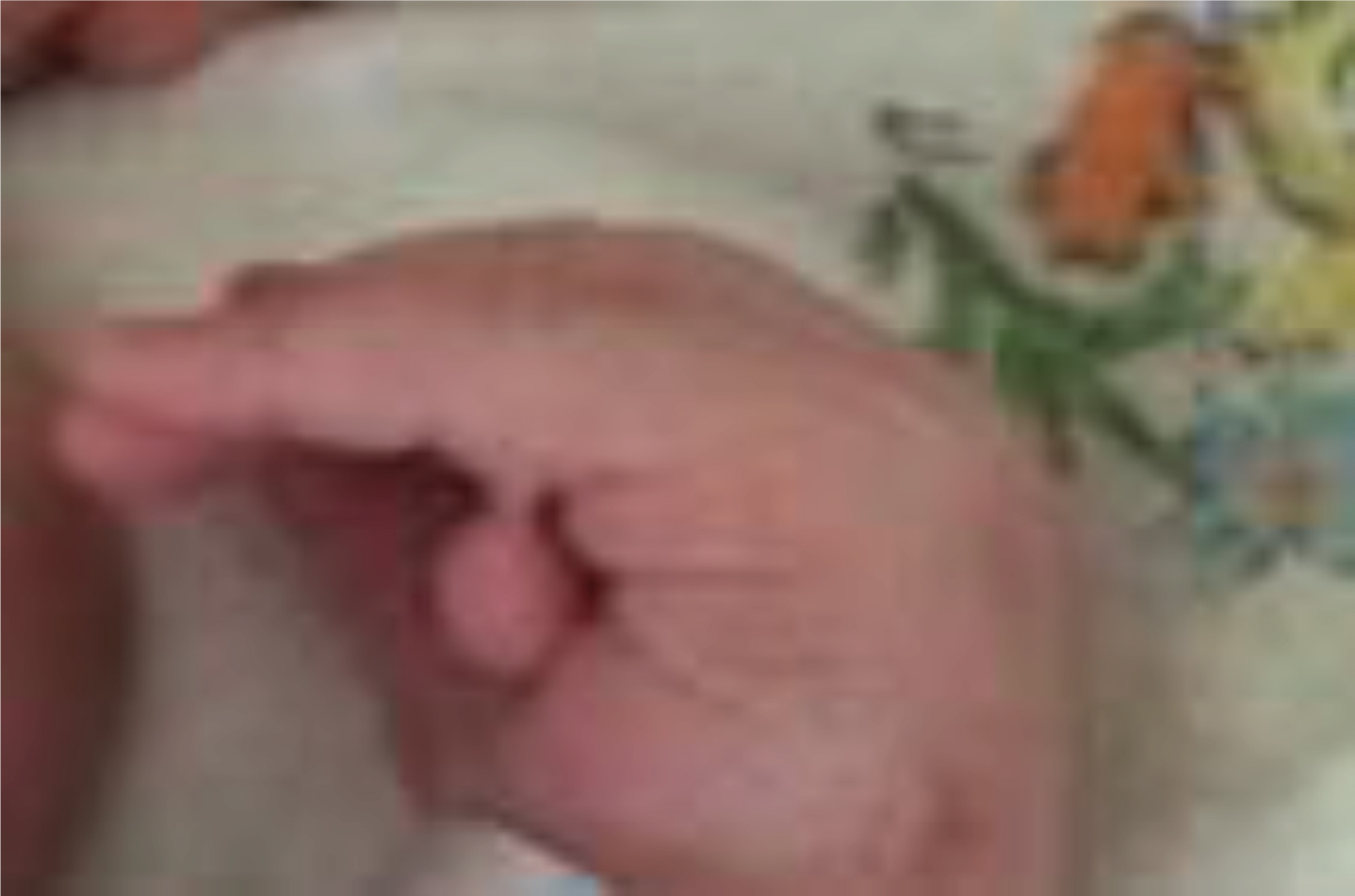
Postaxial polydactyly. Noted at birth.
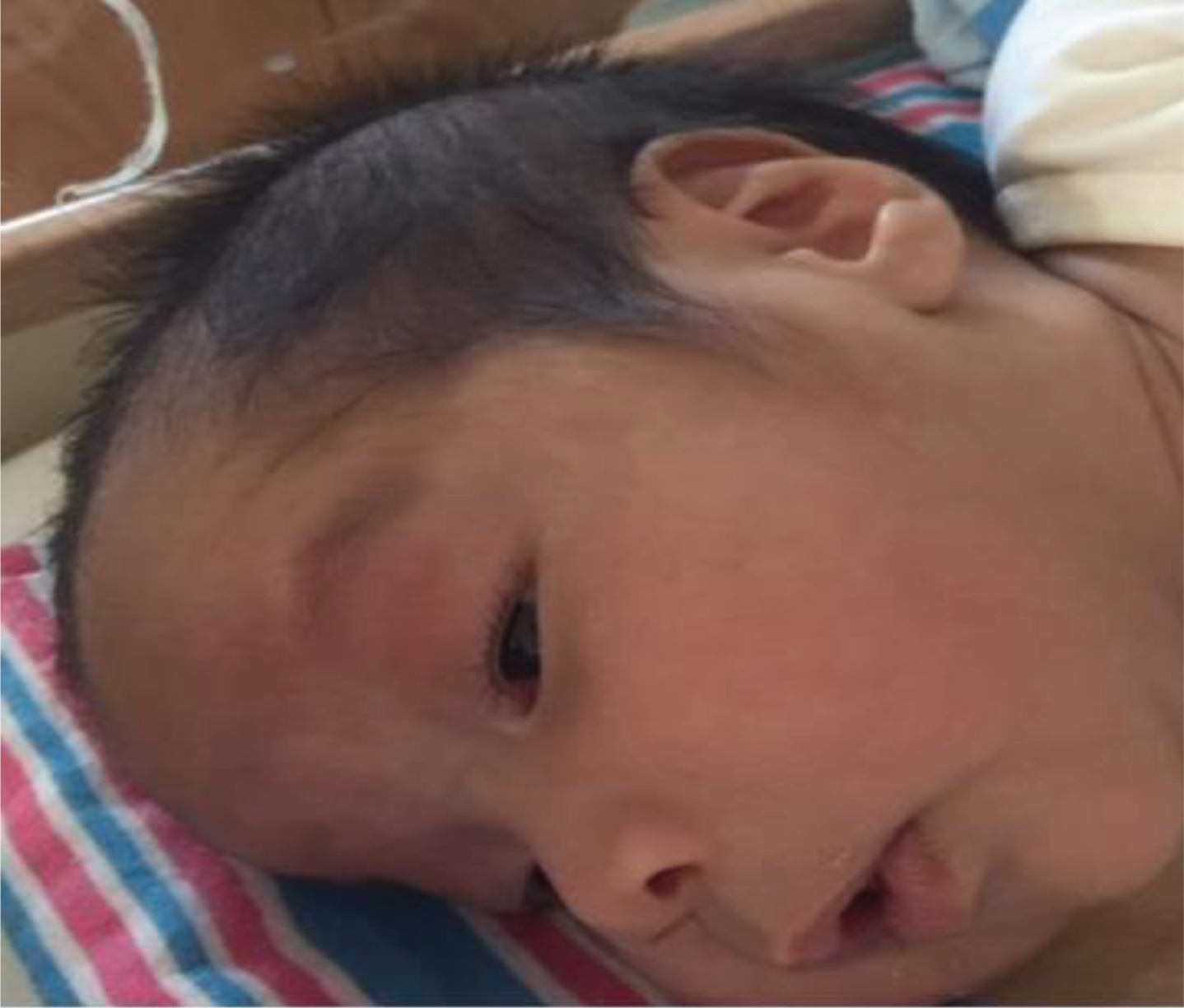
Ptosis on examination. Manifested at around 1 week of age.
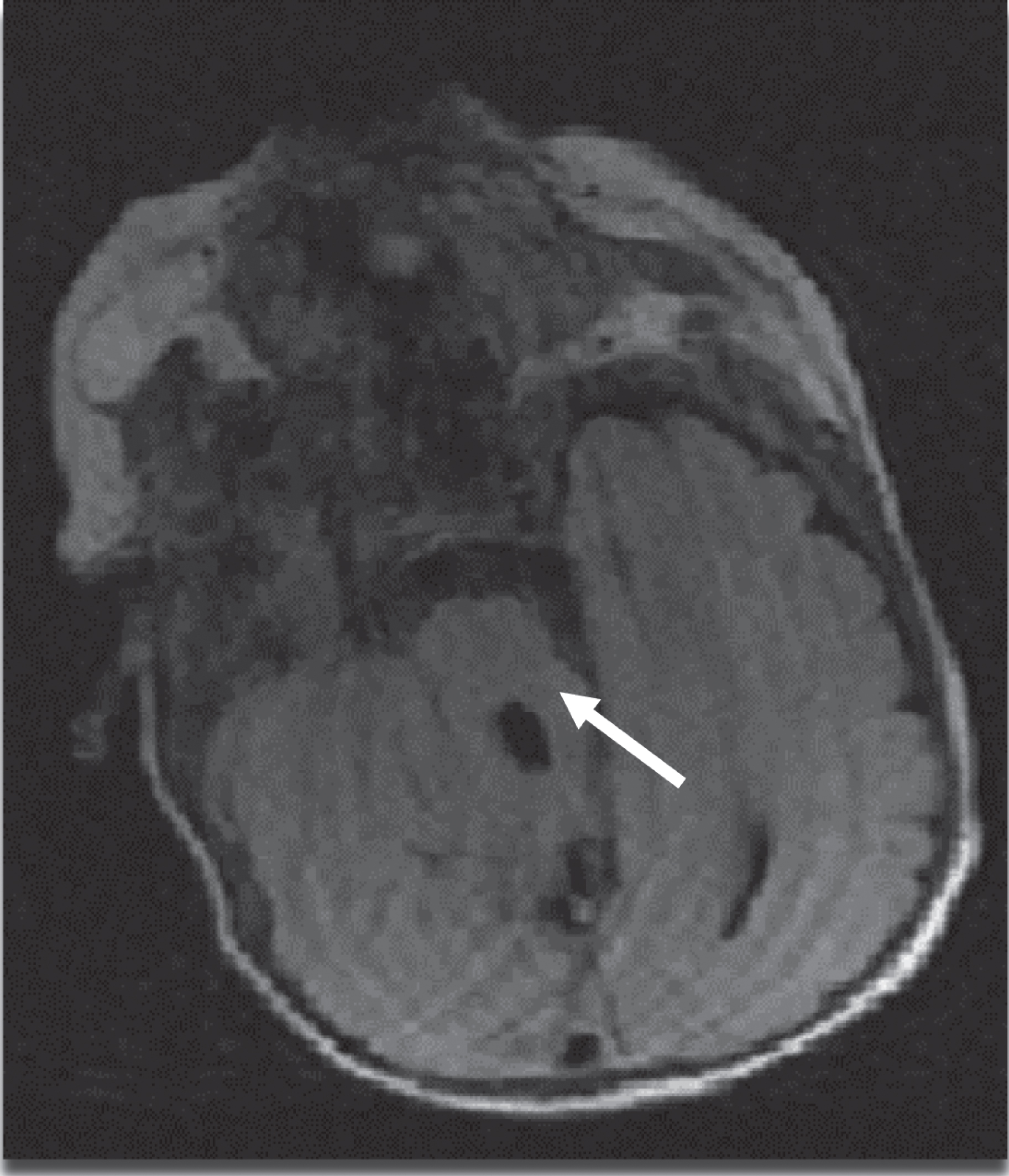
MRI Axial T2 view of brain showing classical pathognomonic molar tooth appearance of mid brain (white arrow).

MRI Brain T1 Sagittal view showing atrophy of the cerebellar vermis (white arrow).
Management is usually symptomatic and multidisciplinary including education programs, physical, occupational, and speech therapy. This may improve the hypotonia, and reduce the delay in achieving motor milestones. Prognosis is favorable for moderate forms of the disease. Management of patients with more severe forms is guarded and usually carried out at a specialized reference center. Differential diagnosis involve Dandy Walker malformation, Senior-loken syndrome, congenital cerebellar hypoplasia, and congenital muscular dystrophy.
The infant in this case is being followed now at the neuro developmental clinic. He has truncal hypotonia with motor delay, and is receiving all the special services. He is taking all feeds orally and is gaining weight with no episodes of desaturations.
The newborn in this case presented initially with only intermittent unexplained episodes of desaturation both at random, and with feeds without any major neurological findings on examination thus delaying the diagnosis. He subsequently developed neurological finding leading to suspected diagnosis with brain neuroimaging and genetic confirmation. This case may aid clinicians to consider JS in infants presenting initially with only unexplained intermittent desaturation and adds to the limited literature on the clinical manifestation in the immediate newborn period.
Conflicts of interest
All authors have no reported conflicts of interest.
Consent
Written informed consent was obtained from the mother of the patient for publication of this case report and images.