
Abstract
Objectives
Microvascular endothelial cell (MVEC) apoptosis is considered to be a key event in the pathogenesis of systemic sclerosis (SSc), an increased expression of endothelin-1 (ET1) is also well recognized in the disease. ET1 is thought to exert deleterious effects on the vasculature by virtue of its known vasospastic, proliferative and fibrotic effects, yet ET1 can act as a survival factor for a variety of cells, including MVEC. The aim of this study is to investigate if ET1 signaling protects SSc-MVECs from apoptosis.
Methods
The expression levels of ET1-receptor genes: Endothelin Receptor Type A gene (EDNRA) and Endothelin Receptor Type B gene (EDNRB), and the effects of selective Endothelin Receptor Type A (ETA) antagonists, selective Endothelin Receptor Type B (ETB), and dual ETA/B antagonist in the presence and/or absence of ET1 on control and SSc-MVEC apoptosis were examined.
Results
Significant increase in the expression of ETA and ETB was noted in SSc-MVECs. Growth factors withdrawal (GFW) resulted in a significant apoptosis that was considerably reduced by the addition ET1. The addition of ETA-receptor antagonists did not affect ET1 anti-apoptotic effects, while the nonselective ETA/B or the selective ETB-receptor antagonists blocked the anti-apoptotic effects of ET1. Finally, an upregulation of the proapoptotic gene BAX after GFW was noted that was normalized by the addition of ET1.
Conclusions
The results suggest that ET1 mediates an anti-apoptotic effect through engaging the ETB receptors in MVECs. Therefore, it appears that selective ETA antagonism may have an advantage over the non-selective ET1-receptor antagonists in SSc vasculopathy, particularly in the early stages of the disease when MVEC apoptosis is rampant.
Keywords
Introduction
Systemic sclerosis (SSc) is a chronic autoimmune disease characterized by progressive vascular dysfunction, immune activation, and extensive tissue fibrosis that is associated with substantial morbidity and mortality (1). There is evidence to suggest that microvascular endothelial cell (MVEC) apoptosis is a primary pathogenic event in SSc. This conclusion is based on the noted MVEC apoptosis in the early stages of SSc (2), and in SSc animal models (3, 4). It is generally accepted that MVEC apoptosis/injury results in vascular dysfunction that is best illustrated by the dysregulated vascular tone control leading to vascular spasm and reduced blood flow.
Endothelin-1 (ET1) is a powerful dose-dependent vasoconstrictor that is believed to be a major contributor to vascular dysfunction and tissue fibrosis in SSc through direct effects on vessels leading to vasoconstriction, hypoxia and ischemia of tissues, and by promoting fibroblast differentiation to myofibroblast (5), leading to enhanced collagen synthesis (6). ET1 is overexpressed in SSc as illustrated by elevated circulating levels and increased tissue expression in the lungs, kidneys, liver and skin (7–8–9–10).
ET1 mediates its biological effects through two receptor subtypes: endothelin receptor type A (ETA), and endothelin receptor type B (ETB). ETA receptors are preferentially expressed in human vascular smooth muscle cells and fibroblasts, where they mediate the vasoconstrictive and proliferative effects of ET1 (11, 12). Whereas, ETB receptors are found primarily in the vascular endothelium, where they mediate vasodilation by stimulating nitric oxide and prostacyclin production (13, 14). Despite the convincing evidence of a potential pathogenetic role for ET1 in SSc, and the significant success of ET1 antagonists (selective and nonselective) in the treatment of certain SSc vascular complications such as pulmonary hypertension and the prevention of digital ulcers recurrences, the precise role of ET1 in vascular dysfunction and MVEC apoptosis remains speculative. Since MVEC apoptosis is recognized as a triggering event in the pathogenesis of SSc vasculopathy and since ET1 signaling through the ETB receptor was shown to provide a survival benefit in rat endothelial cells (15, 16), we hypothesized that the preservation of MVEC-ETB receptor integrity with the use of selective ETA-receptor blockade might minimize MVEC apoptosis. In this study, we demonstrate that ET1 signaling through ETB receptors favors MVEC survival.
Materials and Methods
Drugs and chemicals: the following selective and non-selective ET1-receptor antagonists were used in the study: PD145065, a none selective ETA and ETB-receptor antagonist (17) was obtained from Alexis Corp (Saint Louis, Missouri, USA); BQ788, a selective ETB-receptor antagonist with ETB/ETA affinity of 1/1000 (18) and FR139317 a selective ETA-receptor antagonist with an ETA/ETB affinity of 1/7000 (19, 20) were purchased from Tocris Bioscience (Bristol, UK); and ambrisentan a selective ETA antagonist with affinity for ETA/ETB that ranges between 1/77-4000 (21, 22) was obtained from Gilead Science, Inc.; endothelin 1 (ET-1) was purchased from Sigma (Darmstadt, Germany).
Dermal microvascular endothelial cell isolation: after obtaining informed consent in compliance with the Institutional Review Board of Human Studies, a 5-mm skin biopsy was obtained from affected skin (dorsal forearm) of four patients with diffuse cutaneous SSc of less than 3 years’ duration and from age, gender, and race matched healthy controls. All patients fulfilled the 2013 American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) classification criteria (23). All subjects had skin thickening of the fingers of both hands extending proximal to the metacarpophalangeal joints, and they had Raynaud's phenomenon. None of the subjects included in this study had digital ulcers or pulmonary arterial hypertension at the time of the study. We did not include subjects who were on immunosuppressive or steroid therapy. Microvascular endothelial cells (MVECs) were isolated from the biopsy samples and purified by CD31 magnetic beads as previously described (24) and cultured in Clonetics Endothelial Cell Basal Medium-2 (EBM-2) supplemented with EGM-2-MV growth factors (EGM-2) at 37°C in 5% CO2. The purity of isolated cells was >98%, as determined by flow cytometry analysis using PE anti-human CD31 (BD-Pharmingen, Franklin lakes, NJ, USA). For growth factor withdrawal (GFW) experiments, cells were cultured in DMEM medium without serum and growth factors for 24 h. All studies were performed on cells in the 3th-5th passages.
mRNA extraction and real-time polymerase chain reaction (PCR) detection: total RNA was extracted from cultured cells using the RNeasy Mini Kit according to the manufacturer's nstructions (Qiagen, Valencia, CA, USA) in the presence of DNaseI and quantified spectrophotometrically. RT-qPCR was performed as described previously (25). Briefly, first-strand cDNA synthesis was carried out using an RT-First Strand Kit (SABiosciences, Valencia, CA, USA) according to the manufacturer's instructions. Quantitative PCR was performed with a 7500 RT-PCR System (Applied Biosystems, Grand Island, NY, USA) using Power SYBR Green PCR Master Mix (Applied Biosystems). The differences in mRNA expression between samples were determined using the relative quantification method. The PCR program was conducted at 95°C for 10 min as an initial polymerase activation step, followed by 40 amplification cycles at 95°C for 15 sec and 60°C for 1 min. The cycle threshold (Ct) values of the samples were normalized to the Ct values of the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The fold difference in gene expression of the samples was calculated using the equation 2−∆∆Ct. A list of the PCR primers used for each gene follows: GAPDH forward, 5′-TGCCA AA TATGATGACATCAAGAA-3′, GAPDH reverse, 5′-GGAGTGGGTGTC GCTG TTG-3′; EDNRA forward, 5’-CACTGGTTGGATGTGTAATC-3’, EDNRA reverse, 5’-GGAGATCAATGACCACATAG-3’; EDNRB forward, 5’-TCCCGTTCAGAAGACAGC TT-3’, EDNRB reverse, 5’- CAGAGGGCAAAGACAAGGAC-3’; BAX forward, 5’- AAC ATGGAGCTGCAGAGGAT-3’, BAX reverse, 5’- CAGCCCATGATGGTTCTGAT-3’.
Cell viability assay (MTT assay): cell viability was quantified by the MTT assay, using the Cell Titer Nonradioactive Cell Assay Kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. The values were expressed as percentages over those of controls.
Western blot: Protein detection was carried out with primary antibodies using rabbit monoclonal anti-BAX, rabbit polyclonal anti-ETAR, rabbit monoclonal anti-ETBR (Abcam Inc. Cambridge, MA, USA); mouse monoclonal anti-GAPDH (Santa Cruz Biotechnology, Dallas, TX, USA). Bands were visualized with the corresponding horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) and detected using the enhanced chemiluminescence (Pierce, Rockford, IL, USA), followed by autoradiography
Annexin V staining: cells were incubated with Annexin-V-Fluorescein (green fluorescence) in Hepes buffer containing PI (Propidium, red fluorescence) and examined under fluorescence microscopy using the Annexin-V-Fluos Staining Kit (Roche, Indianapolis, IN, USA). Cells were harvested and processed according to the manufacturer's instructions.
Gene knockdown assay: small interference RNA (siRNA) against human EDNRA and /or EDNRB were purchased from Invitrogen and transfected into MVECs using the Basic Nucleofector Kit for Primary Mammalian Endothelial Cells (Amaxa, Cologne, Germany) according to the manufacturer's instructions. After 36 h, the cells were collected for RT-qPCR assays to examine the transfection efficiency and knockdown efficiency (26).
RT2 profiler PCR array system: the expression levels of 84 apoptosis-related genes were examined using the Human Apoptosis RT2 Profiler PCR array (SuperArray Bioscience). Total RNA was isolated by TRIzol reagent (Invitrogen) and RNeasy mini kit (Qiagen). cDNA was synthesized from 1 μg of RNA using a SuperScript RT II enzyme (Invitrogen). PCR was performed with the RT2 Profiler PCR array system according to the manufacturer's instructions using ABI 7500 (Applied Biosystems).
Statistical analysis: each experiment was run in triplicate. We used unpaired t-test for the comparison of unpaired data. Statistical significance was defined as a p value of <0.05.
Results
ETA and ETB mRNAs expression levels in SSc and control MVECs
The expression levels of EDNRA and EDNRB in SSc-MVECs and control-MVECs were determined by qPCR and by western blots. Significant upregulation of ETRA and ETRB in SSc-MVECs compared to control MVECs were seen both on the messenger RNA (mRNA) and protein levels, particularly for ETRA (Fig. 1).
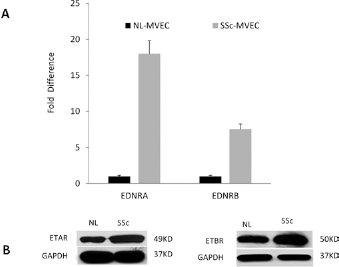
ET receptors expression levels. The expression level of EDNRA and EDNRB, which encodes for ETA and ETB receptors, respectively, in SSc- and control-MVECs, were evaluated by RT-qPCR (A) and by Western blots (B). Overexpression of both ETA and ETB receptors, particularly ETA receptors, was seen in in SSc-MVECs. The graph represents mean ± SD of triplicate experiments. MVECs = microvascular endothelial cells; SSc = systemic sclerosis; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; EDNRA = endothelin receptor type A; EDNRB = endothelin receptor type B.
ET1 protection of MVEC GFW-induced apoptosis
A significant degree of apoptosis was noted in MVEC cultures 24 h following GFW or in response to oxidative stress, especially in SSc-MVECs (Fig. 2A, Supplementary Fig. 1, available online as supplementary material at www.sclerodermajournal.com). Specifically, upon GFW the cell viability was 65 ± 4% in control-MVECs and 49 ± 6% in SSc-MVECs. The addition of ET-1 at 100 nM (a concentration comparable to the effective concentrations used to demonstrate in vitro biologic effects for ET-1) resulted in significant, albeit incomplete, reversal of MVEC apoptosis (cell survival 91 ± 6% in control-MVECs and 75 ± 4% in SSc-MVECs, Fig. 2B), representing substantial ET1 anti-apoptotic effects.
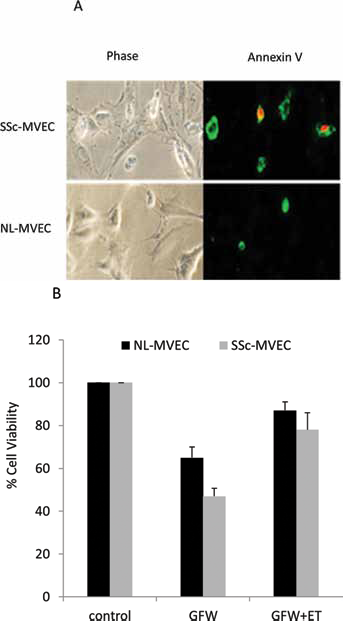
MVEC apoptosis following GFW. Phase contrast image and immunostaining for MVECs (SSc and control) depicting cell apoptosis following GFW, apoptosis was detected by Annexin V stain after 24 hours of growth factor withdrawal (A, annexin stain in green). Normal- and SSc-MVECs were cultured in regular medium (control), with GFW, or with GFW and ET-1 (100 nM) for 24 h. Percent cell viability was calculated using experimental optical density (OD) value/control OD value of triplicate experiments (B). ET1 = endothelin-1; SSc = systemic sclerosis; GFW = growth factors withdrawal.
Selective blockade of ETB, but not ETA, reversed ET1-mediated anti-apoptotic effects. The effect of the selective ETB-receptor antagonist BQ788 (1 µM) was tested by the addition of BQ788 to SSc-MVECs after GFW. Cells were incubated for 24 h before the assessment of cell viability. BQ788 by itself did not affect GFW-induced MVEC apoptosis. However, when BQ788 and ET1 were added to GFW cultures, BQ788 blocked the anti-apoptotic effect of ET1, suggesting that ET1 protective effect is mediated through its binding to ETB receptors (Fig. 3A). Similarly, the addition of the selective ETA antagonist FR139317 (1 µM) or ambrisentan alone did not alter the level of MVEC apoptotic responses. Whereas, when FR139317 or ambrisentan were added with ET1, the anti-apoptotic effect of ET1 was maintained, suggesting that ETA receptors are not involved in ET1-mediated anti-apoptotic effects (Fig. 3B). Furthermore, the addition of the nonselective ET1-receptor antagonist PD145065 at 1 µM to GFW MVEC cultures resulted in reversal of ET1 anti-apoptotic effects (Fig. 3C).
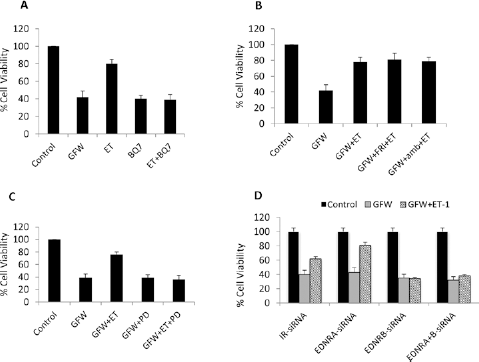
Effects of ET-1 and ET-receptor antagonist on MVEC apoptosis. Twenty-four hours after subjecting SSc-MVECs to GFW, cell viability was 49 ± 6%. The addition of ET-1 resulted in an increase in cell viability to 75 ± 4% (3A). The addition of the selective ETB-receptor antagonist BQ788 (1 uM) blocked the anti-apoptotic effect of ET-1 (A); the data represent percent mean cell viability of triplicate samples ± SD. However, when the selective ETA-receptor antagonists (FR139317 and/or ambrisentan) were added together with ET-1, the anti-apoptotic effect of ET-1 was maintained (B) (data represent percent mean cell viability of triplicate samples ± SD). The addition of the nonselective ETA antagonist PD145065 blocked ET-1 anti-apoptotic effect of ET-1 (C). The protective effect of ET-1 was maintained after transfection of MVECs with Ir- or EDNRA-siRNA, while it was completely lost in cells transfected with EDNRB-siRNA and in cell transfected with both EDNRA- and EDNRB-siRNA (D) (data represent percent mean cell viability of triplicate samples ± SD). ET1 = endothelin-1; SSc = systemic sclerosis; GFW = growth factors withdrawal; EDNRA = endothelin receptor type A; EDNRB = endothelin receptor type B.
ETA and ETB receptor knockdown
To confirm that ET1 signaling through ETB receptor mediates ET1 protective effects, we performed siRNA knockdown experiments followed by examining ET1 effects on MVEC apoptosis using the same experimental design. siRNA knockdown resulted in >95% reduction in the expression level of EDNRA, EDNRB or both (Supplementary data 2, available online as supplementary material at www.sclerodermajournal.com). No effect was noted for IR-siRNA, or EDNRA-siRNA on ET1 anti-apoptotic effect, while EDNRB-siRNA and the combined transfection with EDNRA-siRNA and EDNRB-siRNA blocked ET1 protective effect (Fig. 3D), suggesting again that ETB receptor mediates MVEC survival effects of ET1.
The effect of the selective ETA-receptor antagonist on cell survival is dose dependent
Ambrisentan was added at 1 uM, 10 uM, 100 uM, and 1 mM concentrations together with ET1 to GFW subjected SSc-MVECs. The anti-apoptotic protective effect of ET1 on MVECs was lost with increasing ambrisentan concentrations (Fig. 4). The observation suggests the possibility of loss of ETA selectivity at higher concentrations. The lack of protection at higher doses is not related to the induction of MVEC apoptosis by the higher concentrations of ambrisentan since adding higher concentrations of ambrisentan to MVEC cultures without ET1 did not result in apoptotic response (data not shown).
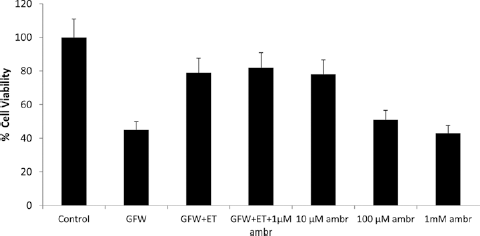
Effect of ambrisentan on SSc-MVEC apoptosis after GFW. The protective effect of ET-1 in the presence of ambrisentan was lost with increasing concentration of ambrisentan suggesting possibly the loss of ETA selectivity with increasing concentrations from 1 µM to 1 mM. ambr = ambrisentan; ET1 = endothelin-1; SSc = systemic sclerosis; GFW = growth factors withdrawal; ETA = endothelin receptor type A.
The molecular mechanism for ET1-mediated anti-apoptotic effect in MVECs
To study the mechanism of ET1-mediated anti-apoptotic effects in MVECs, we performed focused apoptosis gene expression microarray profiling of MVECs after GFW and examined the effects of ET1-, ETA- and ETB-receptor antagonists on gene expression profile. GFW was associated with a significant upregulation of the proapoptotic gene BAX, and the addition of ET1 downregulated BAX gene expression to baseline levels. The addition of ambrisentan maintained ET1 downregulation of BAX, while the nonselective-receptor antagonist PD145065 inhibited ET1 effect on BAX expression (Fig. 5). These observations further suggest that the ET1 effect is mediated by ETB receptors. No effect was noted for either ambrisentan or PD145065 on BAX expression when they were added alone to GFW subjected MVEC cultures without the addition of ET1, which argue for a specific role for these agents in blocking ETB-mediated signaling pathways.
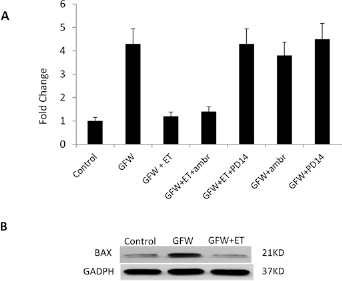
Expression level of the proapoptotic gene BAX. (A) BAX mRNA expression level measured by the Human Apoptosis RT² Profiler™ PCR Array. Four-fold increase in the expression level of BAX mRNA was noted following GFW. The addition of ET-1 inhibited the upregulation of BAX mRNA expression. Addition of ambrisentan maintained ET-1 effect, while addition of the nonselective ETA/ETB antagonist PD145065 completely blocked ET-1 effect on BAX expression. No effects were noted for the antagonists alone. (B) Effects of GFW and ET1 on BAX expression levels examined by western blot. Equal amounts of protein were uploaded on western blot gels. ET1 = endothelin-1; GFW = growth factors withdrawal; ETA = endothelin receptor type A; ETB = endothelin receptor type B.
Discussion
The endothelin system was first identified in the late 1980s from cultured endothelial cells (27). Four isoforms are currently recognized; however, endothelin-1 (ET-1) is the most widely expressed member of the endothelin family that was originally described as a very potent vasoconstrictor. Recent data suggest that ET-1 is a true pluripotent cytokine. It can exert several vascular effects, including vasoconstriction, cellular proliferation and hypertrophy, as well as non-vascular effects, notably stimulation of cardiac hypertrophy, tissue fibrosis and inflammation. There are two distinct ET receptors, ETA and ETB, which can mediate opposing effects (28). In normal physiology, ETA receptors are expressed on smooth muscle cells, while ETB receptors are present on the endothelium and to a lesser extent on smooth muscle cells. Activation of both ETA and ETB receptors on smooth muscle cells leads to vasoconstriction, while activation of ETB receptors on endothelial cells leads to vasodilation that is mediated by stimulation of nitric oxide and prostacyclin production. In terms of cell survival, it appears that ETB blockade induces cell apoptosis of glioma cells and melanoma cells (29, 30), suggesting a protective effect of ET1 through ETB signaling. Sakai et al recently demonstrated that ET-1 has an anti-apoptotic effect on cultured pulmonary artery smooth muscle cells (PASMC) that is mediated by ETB receptors. Theoretically, in the case of PASMC, inhibition of ETB using dual ETA+B antagonist would be preferential to ameliorate vascular proliferation by induction of PASMC apoptosis (31).
Numerous clinical studies have documented the involvement of the ET system in a variety of vascular and fibrotic disorders. In SSc, increased ET-1 production, particularly in diffuse SSc, pulmonary fibrosis, pulmonary arterial hypertension, and SSc renal crisis have been reported (32). In the skin, increased dermal ET-1 expression is seen in involved and uninvolved skin, predominantly in the superficial vessels, particularly in the early active stages of the disease (33). In addition, significant increases in ET-1 binding sites with upregulation of both ETA and ETB receptors in the microvessels, epidermis, and hair follicles of SSc skin are seen.
MVEC apoptosis was first described on ultrastructural examination of SSc skin biopsies in the early inflammatory stages of the disease (34). It was later noted in the University of California at Davis (UCD) lines 200/206 chickens that spontaneously develop a systemic disease that closely resembles human SSc (35). MVEC apoptosis is considered to be a primary and early pathogenic event in SSc that leads at later stages to vascular dysfunction, obliterative microvascular disorder and arteriolar fibroproliferative vasculopathy.
In this study we employed growth factors withdrawal (GFW) to induce MVEC apoptosis. Endothelial cultures in serum withdrawal conditions is an established technique for apoptosis induction and has been extensively used to induce apoptosis in nontransformed cells because of its reproducible effects. GFW induces apoptosis by activation of mitochondrial cytochrome C release, increased production of reactive oxygen species, and by induction of conformational changes in BAX (36–37–38). In other experimental models, GFW-associated apoptosis was shown to involve caspase-1 activation (39).
In this study, we provide experimental evidence for the role of ET-1 in mediating an anti-apoptotic effect through ETB-receptor signaling in MVECs. First, we demonstrate that ET-1 rescues MVECs from GFW-induced apoptosis. Next, we show that both non-selective ET and selective ETB-receptor antagonists block ET-1 anti-apoptotic effects, while selective ETA antagonists maintain ET-1 anti-apoptotic effects. Furthermore, siRNA knockdown of ETB, but not ETA, prevented ET-1 anti-apoptotic effect. This observation is supported by data demonstrating ET-1 ability to protect rat endothelial cells and human umbilical endothelial cells from apoptosis induced by various apoptotic stimuli including serum deprivation (40). Consistently, our findings in this study indicate that ET-1 endothelial protective effects can be extended to SSc-MVECs.
The second important finding in our study is the increased mRNA expression of both EDNRA and EDNRB in SSc-MVECs compared to control-MVECs, with surprisingly more increase in EDNRA expression. In contrast, studies that used whole fibrotic lung tissues from SSc patients showed downregulation of ETA expression levels, while ETB levels were slightly increased (41). Moreover, there is evidence for downregulation of ETA in skin fibroblasts from patients with SSc (5). Our data demonstrate an upregulation of ETA gene expression in MVECs, which normally does not express ETA to this extent. Therefore, it appears that increased ET-1 and ETA-receptor expression levels signify potential amplification of ETA signaling in SSc-MVEC. The mechanistic reason for ETA upregulation in SSc-MVECs is not known. Nonetheless, the mechanism for ETB-mediated anti-apoptotic effect is likely to be related to the downregulation of the pro-apoptotic BAX gene. These findings underscore the role of ETB signaling as a protective mechanism against MVEC apoptosis.
Receptor selectivity in this, and other studies, is based entirely on an in vitro system, and the uncertainty of in vivo consequences of receptor specificity is related to receptor dimerization (42) and the interaction between the two receptor types “cross talk” due to functional compensation of the unblocked receptor (43, 44). Nevertheless, our data provide experimental evidence for the advantage of salvaging ETB-signaling pathway by using selective ETA antagonists as a logical approach to treatment of SSc-vascular disease, particularly in the early stages of the disease when MVEC apoptosis is rampant. Further studies, especially in vivo studies, are needed to fully explore the role of endothelin-1, ETA, and ETB receptors in endothelial dysfunction in SSc. The impact of the endothelial protective effect of ET-1 noted in this study can only be fully appreciated by examining this effect in an in vivo disease model. Moreover, we believe that the role of ET-1 receptors in different target cells at different stages of the disease should be re-examined.
In conclusion, our findings point to the potential superiority of selective inhibition of ETA over non-selective inhibition in the early stages of SSc vascular disease. The data propose this advantage may be related to prevention of MVEC apoptosis and the subsequent events that follow the initial injury in the vasculature.
Footnotes
Financial support: This study was sponsored by Gilead Sciences, Inc. as an investigator-sponsored research project. Gilead had no role in the study design or in the collection, analysis, or interpretation of the data, or in drafting the manuscript. Gilead reviewed this publication for the limited purposes of maintaining confidential information and preserving its intellectual property rights.
Conflict of interest: None of the authors has financial interest related to this study to disclose.