
Abstract
In recent years, the homeobox gene superfamily has been introduced as a master regulator in downstream target genes related to cell development and proliferation. An indispensable role of this family involved in organogenesis development has been widely demonstrated since expression of Six family led to a distinct increase in development of various organs. These functions of Six family genes are primarily based on structure as well as regulatory role in response to external or internal stimuli. In addition to these roles, mutation or aberrant expression of Six family plays a fundamental role in initiation of carcinogenesis, a multistep process including transformation, proliferation, angiogenesis, migration, and metastasis. This suggests that the Six superfamily members can be considered as novel target molecules to inhibit tumor growth and progression. This review focuses on the structure, function, and mechanisms of the Six family in cancer processes and possible strategies to apply these family members for diagnostic, prognostic, and therapeutic purposes.
Introduction
A gene network that has a role in determination of cell fate includes the dachshund (dac), twin-of-eyeless (toy), eye absent (eya), teashirt (tsh), and sine oculis (So). The sine oculis homeobox 1 (Six1) gene is a homologue of the Drosophila sine oculis (So) in vertebrates. There are 6 members that have been classified into 3 subgroups, Six1/Six2 (So), Six3/Six6 (Optix), and Six4/Six5 (Dsix4), characterized by a Six-type homeodomain (HD, 60 amino acids) and Six domain (SD, 110-115 amino acids) (1). Up to now, Six family genes have been identified in various species ranging from lower invertebrates including nematodes to higher mammals like humans (2). Several studies have demonstrated that Six family genes play an important role in organogenesis (3) and diseases (4). Among these, Six homeobox 1 (Six1), the most extensively studied, is involved in the development of many tissues and organs, such as craniofacial structures, sensory organs, the auditory system, kidney, and muscle. In addition, the role of Six1 in tumorigenesis has been highly attention-getting. Numerous studies have documented that Six1 is involved in the occurrence of various human cancers, including breast cancer (5), ovarian cancer (6), cervical cancer (7), hepatocellular carcinoma, rhabdomyosarcoma (8), Wilms tumors (9), and colorectal cancer (10). In this review, we summarize the mechanism of the Six1 family, mostly Six1, in development of different diseases, including tumorigenesis.
Homeobox genes
Homeobox genes were first identified in Drosophila due to their ability to cause homeotic transformations, that is, the formation of body parts in inappropriate contexts or locations. Ectopic expression of the antennapedia gene in Drosophila leads to a set of middle legs formed in position of antennae (11). As in this discovery, in all eukaryotic species, including humans, homeobox genes have been observed. The human genome contains at least 200 homeobox genes (12). Because of polarity, and because overall cellular identity and body patterning are influenced by homeoproteins, it is typical for individual superfamily members to modulate the transcription of many target genes to utilize pleiotropic effects in the cell and execute entire programs of organogenesis (13). Members of the Six gene family belong to the superfamily of homeobox genes; by encoding transcription factors through repression and activation of a diverse range of downstream target genes, they have an essential role in development (14). The homeoproteins of these genes control many cellular processes, including cell adhesion, differentiation, cell shape, proliferation, and apoptosis.
Structure of Six homeoprotein
Conserved 61‐amino acid DNA‐binding motif known as the HD is a hallmark of superfamily transcription factors identification (15). The HD helix‐turn‐helix motif is responsible for binding homeoprotein to DNA in a sequence‐specific manner (13). The HD of Six family homeoproteins differs from classic HDs. Because of the lack of conservation in arginine residue at position 5 and glutamine residue at position 12 in helix 1, serine or threonine is replaced in the Six type HD (1). Replacement of residues 5 and 12 make the N‐terminal region of the Six HD have a novel structure that differs from most HDs. In addition to the conserved Six HD, the Six family contains a second, highly conserved domain that is located N‐terminally from the HD. The SD, which refers to this N‐terminally conserved domain, was initially thought to put in to both protein interactions with cofactors and DNA binding (1). The activating transcription elements in Six2 and Six4 have been related to C‐terminal domains of them. It is shown that some of the Six family members do not always require eya to activate transcription as they are bona fide transcription factors (16). Six5 is similar to Six4 and is necessary for its ability to activate transcription and possibly contain an activation domain in its C‐terminus (17). Intrinsic activation domains have not been found in the other Six family members (Six1, Six3, Six6), presumably requiring these Six proteins to rely more heavily on interactions with cofactors (16). Although Six1 HD exhibits a DNA sequence preference, it is substantially different from that exhibited by the full-length protein. It is concluded that outside regions of the Six1 homeodomain participate in DNA binding either through direct DNA contacts or indirectly by enabling stabilization of structure (18).
Six family and cancer
Several members of the Six family have been implicated in the process of proliferation that leads to differentiation, and this proliferative function may engage in tumorigenesis. Aberrant expression of Six family members has been observed in cancers: Six3 in extraskeletal myxoid chondrosarcomas (19), Six5 in borderline ovarian tumors (20), Six6 in acute T‐cell leukemias (21), and Six1 in breast (22), ovarian (6), cervical (23), and hepatocellular carcinomas (24), and pediatric malignancies such as rhabdomyosarcomas and Wilms tumors (25). Besides, amplification of the 14q23 locus, including Six1, Six4, and Six6, may have caused breast cancer (22). Thus, in almost all the aforementioned tumor types, expression of Six family members is inappropriate, which suggests that their aberrant expression may promote tumor progression and/or tumorigenesis. The precise role of most Six family members in cancers has not been well-determined.
Related mechanisms involved in tumorigenes
Epithelial-mesenchymal transition
Transformation of adherent epithelial cells into invasive mesenchymal cells can cause cancer cell formation. The role of epithelial-mesenchymal transition (EMT) steps during development is well-defined, but the association of EMT with cancer is less clearly determined. Based on the functional consequences and biological context, there are 3 different subtypes of EMT, including Type I (developmental), Type II (fibrosis and wound healing), and Type III (cancer) (26). In cancer, features of EMT have been observed in breast (27), ovarian (28), colon (29), and esophageal (30) cancer models. Changing apicobasal polarity (31), disintegration of tight junctions (32) and adherent junctions, and cytoskeletal changes can cause oncogenic EMT (33). The mechanism of EMT for tumor progression and aggressiveness is shown in Figure 1. Transforming growth factor-β (TGF-β) (34), Wnt (35), Snail/Slug (36), Twist (37), and Six1 (38) are some inducers of EMT in cancer cell lines that have been identified. It has been shown that overexpression of Six1 upregulates mesenchymal-related genes and downregulates epithelial-related genes, which suggests that EMT is induced by Six1 overexpression (39).
The Six family of homeodomain transcription factors, specifically Six1, Six2, and Six4, are important during developmental EMT. In breast cancer patients, increased expression of Six1 correlates with poor prognosis. Elevated levels of Six1 in patients cause increased lymph node involvement, shortened time to relapse and metastasis, and decreased overall survival (40). In breast cancer, misexpression of the Six1 developmental EMT regulator accelerates tumor progression by inducing a similar EMT program in tumor cells. Transforming growth factor-β signaling induces EMT and is required for intravasation, extravasation, and bloodborne metastasis (41). One of the important pathways in numerous homeostatic processes is TGF-β signaling, which plays a significant role in tumorigenesis (42). The normal function of TGF-β during organogenesis can be parallel to its effect in cancer, similar to the other developmental pathways implicated in cancer. For example, EMT is required for cardiac valve formation that TGF-β signaling can induce during development, and similarly, an EMT is induced by treatment of numerous cancer cell lines with TGF-β (43). Significantly, TGF-β signaling can be both tumor suppressive and tumor promotional depending on the context of activated TGF-β signaling pathway (44). In breast cancer, early lesions occur typically by growth-inhibiting TGF-β, which highlights its tumor suppressive activity (45). Nevertheless, in later stages of breast cancer, resistance occurs to the growth inhibitory activity of TGF-β (46), and in its place, TGF-β signaling promotes metastatic progression through multiple mechanisms including EMT induction (47). This switching mechanism of TGF-β signaling from tumor suppressive to prometastatic is not well-determined. Transforming growth factor-β signaling initiates through cell-surface serine-threonine kinase type II and type I receptors. Transforming growth factor-β binding to TβRII (TGF-β type II) receptor triggers its association with the TGF-β type I (TβRI) receptor (48). TβRI is phosphorylated by TβRII. TβRI activation induces phosphorylation of Smad2, Smad3 and receptor-associated (RA) Smads. RA-Smads bind Smad4 which leads to make the complex. (48). On the other hand, In HKc/DR (human keratinocytes toward a differentiation-resistant) with Six1over expression, Smad-independent pathways of TGF-β signaling can be involved in Six1-mediated EMT through TGF-β receptor type II (TβRII)- mitogen-activated protein kinase (MAPK) pathway (39). In addition, TGF-β can activate Jun N-terminal kinase (JNK) (49-50-51), extracellular signal-regulated kinase (ERK) (52), p38 mitogen-activated protein kinase (p38 MAPK) (53), and Akt (54). One of the required elements of persistent Six1-induced EMT and TβRII overexpression is p38 MAPK activation. In HKc/DR, when Six1 is overexpressed, there is malignant conversion and increased cancer stem cell (CSC)-like population. Thus, it can be concluded that through MAPK activation, overexpression of Six1 promotes EMT, CSC properties, and malignant conversion in HKc/DR (39). TβRI is a target of Six1 and its upregulation causes TGF-β signaling activation, which leads to inducing EMT properties. Interestingly, increased TβRI expression is not sufficient to promote experimental metastasis, which proposes in vivo evidence that Six1 overexpression is requisite to switch TGF-β signaling to the prometastatic phenotype. It is demonstrated that induction of EMT cannot induce experimental metastasis. Together, these results demonstrate other activation of TGF-β signaling mechanism in which TβRI is identified as a new target of Six1, as well as implication of this gene as a determinant of TGF-β function in cancer, like breast cancer (38).
Activation of Notch2 signaling pathway depends on cell membrane-bound ligand. One of the reasons for upregulation of both Notch2 and Six1 transcriptional deviation is exchanging preinvasive to invasive adenocarcinoma progression by promoting epithelial-mesenchymal transition and nuclear atypia. Smad3 and Smad4 perform as the intracellular signal transducers in intracellular signaling pathways that lead to EMT activation by Notch2 and Six1, in which upregulation of Notch2 leads to Six1 transactivation in various cancers (55, 56). In order to subclassify lepidic-predominant invasive adenocarcinoma, which is practical to predict the prognostic outcome of patients with lepidic-predominant invasive adenocarcinoma, upregulation of Notch2 and Six1 seem to be significant (57).
The EMT inducers can be microenvironmental factors, including TGF-β and Wnt agonists, as well as the E-box-binding transcription factors Twist, Snail, and ZEB, zinc-finger enhancer transcription factors (ZEB1 and ZEB2, encoded by the ZFHX1a and ZFHX1b genes). They are collectively sufficed for EMT induction by regulating cytoskeletal, cell polarity, cell adhesion, and apoptosis-regulatory genes (58, 59). ZEB1 is induced by a variety of transcription factors including NF-kB, Twist/Snail (acting in concert), TCF4, and LBX1 (60). Six1 can activate transcription of the ZEB1 promoter significantly. On the ZEB1 promoter oligonucleotide that contains 2 tandem Six1 binding sites, the Six1 protein-DNA complex can be effectively abolished by GRHL2 (Grainyhead-like 2) protein (61).
Cell cycle interference
Six1 transcriptional targets, including cyclin A1, cyclin D1, ezrin, and c-myc, have a role in cell growth and proliferation. Overexpression of Six1 leads to an abating of the DNA damage induced by G2 cell cycle checkpoint. Six1 links to the cell cycle as well as to tumor progression (62). Diseases associated with G2 checkpoint control, such as ataxia telangiectasia, Li Fraumeni (63), Bloom syndrome (64), and Fanconi anemia, all reveal susceptibility of cancer. Six1 is overexpressed in a large proportion of breast cancers. Three independent Six1 regulation mechanisms have been revealed in the cell cycle, via transcription, phosphorylation, and degradation. Protein phosphorylation regulates a number of homeodomain-containing transcription factors including Csx/Nkx2.5, Cut, Pit-1, Oct-1, and Drosophila engrailed. Several kinases, including protein kinase casein kinase II (CK2), protein kinase C (PKC), and protein kinase A, can phosphorylate homeodomain-containing proteins; e.g., CK2, a serine/threonine kinase that is ubiquitously expressed, has been shown to phosphorylate transcription factors encoded by Csx/Nkx2.5 (65), Cut (66), Hoxb-6 (67), and Engrailed (68) homeobox genes. Overexpression of Six1 may reduce the DNA damage-induced G2 cell cycle checkpoint in mammary carcinoma cells (62). CK2 phosphorylates Six1 and G2/M is arrested in the same cell type by inhibitors of CK2 (69). In keratinocytes, fibroblasts, and neuronal cells, there is a similar G2/M arrest (70). It has been reported that in response to DNA damage, Six1 is a target for CK2 in cell cycle control at the G2/M transition. This protein (Six1) holds 7 CK2 sites, including 2 PKC consensus sites, and 5 possible cdc2 sites. Mammalian Six1-Six1 and Six9 have conserved N-terminus, at the very end of the Six domains in a region significant for both protein-protein interactions and DNA binding (17, 71). A key role for CK2 in controlling their functions is based on the number of conserved CK2 phosphorylation sites in the Six class members. The activity of the cyclin-dependent kinases (CDKs) that catalyze the ordered transitions from one phase of the cell cycle to the next can be modulated by internal and external signals. A second A-type cyclin is Cyclin A1, which binds CDK2 and is a critical factor in cell proliferation, survival, DNA repair, and angiogenesis (72, 73), greatly expressed in ovarian, testicular, and endometrial cancers. This cyclin is overexpressed in acute lymphoblastic and myeloid leukemias (74, 75), while Six1 overexpression is implicated in the mechanism of malignant transformation of immortalized, nontumorigenic mammary epithelial cells, which involves inappropriate upregulation of cyclin A1 in the adult mammary gland. Observations show that Six1 is critical for invasion and metastasis in ovarian and rhabdomyosarcoma in tumor progression that supports such a role of Six1 (8). The association of Six1 with increased stage as well as poor prognosis in both ovarian and hepatocellular carcinoma has been identified (6, 24). Cyclin A1 also mediates autocrine expression of vascular endothelial growth factor (VEGF) in support of retinoblastoma (Rb) gene- and androgen-dependent pathways in prostate cancer. Not only increased CDK activities may be due to the increased cyclin A1 expression, but also altered expression of CDK inhibitors such as p27, p21, and CDK phosphorylation. There is a physiologic interaction between cyclin A1 and Rb protein. Initially, cyclin A1 interacts with Rb protein; after that, cyclin A1 regulates VEGF expression in cooperation with Rb-dependent pathways. In addition, androgen-mediated pathway, especially androgen receptor (AR), a member of nuclear receptors, is important for the action of cyclin A1, and Rb protein clearly acts as a bridge between cyclin A1 and AR. Cyclin A1 is unable to mediate VEGF expression in the absence of a functional Rb, although AR is present (76). The schematic of this mechanism is shown in Figure 1.
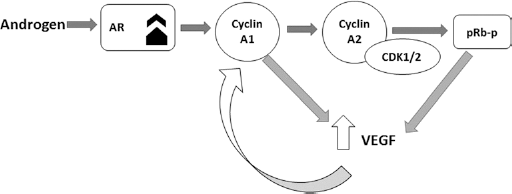
Molecular mechanism of androgen receptor (AR) in regulating vascular endothelial growth factor (VEGF) expression in cooperation with Rb-dependent pathways. CDK1 = cyclin-dependent kinase 1
Ezrin, which can control cell proliferation, survival, and motility, is another transcriptional target of Six1 that plays a role in the metastasis of mammary and pancreatic adenocarcinoma and osteosarcoma (77). In vivo functional studies found that ezrin, the actin filament-plasma membrane linker, and the Six-1 transcription factor are 2 essential elements that affect the metastatic fate of rhabdomyosarcoma. Ezrin acts as a link between the plasma membrane and the actin cell cytoskeleton. However, it also has a role in signal transduction pathways included by protein kinase A, Rho, phosphatidylinositol 3-kinase/Akt, MAPK, and Src. Six1 binds to an oligonucleotide sequence located at ezrin promoter and activates the transcription of genes encoding ezrin (8). Therefore, this complex can be a predictor of metastasis in cancer (78).
One of the active factors subsequently identified in various animal and human tumors is c-myc proto oncogene. Cell cycle protein expression is modulated while c-Myc mediated cell transformation. Expression of Myc or activation of conditional MycER chimeras lead to increase in the level of cyclin A and cyclin E mRNA. While activation of cyclin A expression occurs as a result of Myc expression, growth factor is an independent connector of cyclin A and cdk2 with the transcription factor E2F, which in turn is associated with an increase in E2F transcriptional activity. However, high decreases of the expression of cyclin D1 in G1 phase have been seen in Myc-transformed cells (79).
One of the downstream targets of ERKs is c-Myc. Overexpression of Six1 mRNA has been observed in half of primary breast cancers, and in a high percentage of metastatic lesions (22). One of the mechanisms that Six1 uses for expanding the tumor initiating cell population is activation of mitogen activated protein kinase/ERK signaling.
In human epidermal growth factor receptor 2 (Her2)-enriched and luminal B breast cancers, the highest levels of Six1 mRNA expression were detected. Expression of Six1 has been identified in all breast cancer subtypes (80).
One of the most commonly overexpressed oncogenes in cancers is cyclin D1. Six1 can target the gene encoding the cyclin D1 and led to upregulation of cyclinD1 and promote cell cycle progression and proliferation in pancreatic cancer cells (81).
One of the basic mechanisms that restrict key cell cycle modulators expression in order to appropriate cell cycle stages is ubiquitin-mediated proteolysis.
The ubiquitous component of eukaryotic cell membrane is sphingolipids. Tabasinezhad et al (82) studied the role of this component in tumor progression in their review article thoroughly. Ubiquitin-mediated proteolysis regulates Six1 homeoprotein through anaphase-promoting complex APCCdh1 in a cell cycle-specific manner. Six1 stabilization can occur as a result of a deletion in N-terminus or the C-terminus and led to degradation of Six1 by APCCdh1. Thus, there is possibility that Six1 N-terminus has a new motif that is vital for Cdh1 binding, while C-terminus contains an additional element that can bind to the core of APC (83). The activation of DR4 and DR5, the death receptors, in response to the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas ligand (FasL) following activation of Fas/CD95 can initiate the signaling of apoptosis pathway.
Although Six1 could cause TRAIL resistance, it has slight effect on FasL sensitivity (84). Thus, TRAIL resistance, a common phenomenon in metastatic cancers, may be represented by expression of Six1, which may lead phosphorylation protection against both TRAIL-mediated and FasL-death receptor pathway (85).
Role of microRNAs in the Six Family
microRNAs, a class of noncoding RNAs, play a potential role as diagnostic and prognostic biomarkers in cancers. A recent study showed miR-204 involved in gastric cancer metastasis by post-transcriptional repression of SIRT1 (86). It regulates Six1 expression via direct binding its 3’-UTR. Knockdown of Six1 expression by siRNA could downregulate miR-204, which led to partial abatement of the proliferation and invasion of cells (87).
miR-30b can regulate migration and invasion in metastasis. In primary CRC specimens, miR-30b was downregulated significantly compared with normal tissues, and was much higher compared with liver metastasis tissues. In vitro experiments revealed that compulsory expression of miR-30b inhibited CRC cell migration and invasion by altering Six1 gene expression. Furthermore, an inverse correlation between the expression of miR-30b and Six1 in primary CRC specimens and liver metastasis has been observed (88).
miR-185 is another anticipated microRNA that has a role in Six1-mediated tumorigenesis. In esophageal squamous cell carcinoma, it is shown that after transfection of miR-185 the expression of a Six1 downstream gene, cyclin A1, decreased, which may inhibit proliferation, migration, and invasion through targeting Six1 (89). Furthermore, decrease of miR-185 expression leads to an increase in Six1 expression levels.
The miR106b-25 cluster includes miR-106b, miR-93, and miR-25. It is highly conserved in vertebrates and resides in the 13th intron of the MCM7 gene (Chr7).
Overexpression of Six1 can lead to a switch in TGF-β signaling from tumor suppressive to tumor progressive feature by targeting the Smad-7, TGF-β inhibitor, as well as activating TGF-β signaling pathway. Overexpression of miR-106b-25 is sufficient to induce EMT characteristics and tumor-initiating cells, and is necessary for Six1 to mediate these phenotypes. There is a correlation between the expression of miR-106b-25 and both Six1 and TGF-β signaling pathway in breast cancer tissues. Six1/miR-106b-25 axis is vital for activation of TGF-β signaling in breast cancer (90). The correlation between Six1 and multi potent factorial in cancer is summarized in Figure 2.
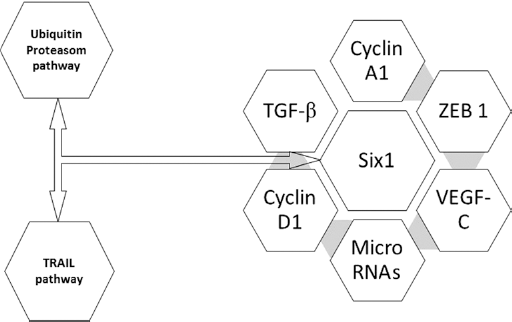
Interaction of Sineoculis homeobox homolog 1 (Six1) with potent signaling molecules involved in tumor progression. TGF-β = transforming growth factor β; VEGF-C = vascular endothelial growth factor C; ZEB1 = zinc finger E-box-binding homeobox 1
Six1 and drug resistance of cancer
Six1 level was associated with resistance to paclitaxel in breast cancer cells. On the contrary, knockdown of endogenous Six1 by siRNA led to sensitization of breast cancer cells to paclitaxel (91). Besides paclitaxel treatment effects, cellular events such as mitotic arrest and apoptosis occurred (92). The data showed that compulsory overexpression of Six1 inhibited paclitaxel-induced apoptosis in MCF-7 cells. Six1 may be involved in paclitaxel resistance through effects of paclitaxel on apoptosis in breast cancer cells. In addition, clinical data showing the expression of Six1 and paclitaxel sensitivity are related clinically. This is in parallel with previous studies reporting that in breast cancer, overexpression of Six1 showed a relationship with poor prognosis (80). This finding suggested that Six1 may play a role in the responsiveness of breast cancer cells to paclitaxel (91). Recombinant TRAIL and/or agonistic monoclonal antibodies are being investigated in phase I clinical trials against some solid malignancies, like ovarian cancer. Since Six1 mRNA overexpression was observed in 63% of late-stage ovarian carcinomas and led to TRAIL resistance, it is possible that Six1 levels in patient tumors will impact the result of these clinical trials (6). Six1 may deliver a new therapeutic target for treatment of breast cancers that are resistant to paclitaxel treatment.
Conclusion
Homeobox superfamily transcription factors have pivotal roles in the development of numerous organs and control processes such as proliferation, apoptosis, migration, and invasion. The Six family is widely expressed in different tissues and organs and levels of these transcription factors during fetal development to adulthood vary, suggesting that there is probably a critical promoter to preserve its basic expression in various tissues. The Six family could regulate target genes by recruiting tissue-specific or stage-specific co-repressors or coactivators.
Overexpression of the Six1 homeoprotein has been found in several malignancies, such as breast, rhabdomyosarcomas, hepatocellular carcinomas, ovarian, and Wilms tumors. Recent evidence determined that Six1 is involved in cellular migration and invasion during embryogenesis and in breast cancer via a process that may include an EMT, which was accompanying by increased TβRI expression and activation of Smad-dependent TGF-β signaling. Since Six1 regulates progression of tumor globally, blockage of the Six1 transcriptional complex might be a powerful method helpful in treatment of many cancers.
Footnotes
Financial support: No financial support was received for this submission.
Conflict of interest: None of the authors has conflict of interest with this submission.