
Abstract
Context
Male patients with congenital adrenal hyperplasia (CAH) may develop bilateral testicular adrenal rest tumours (TARTs). These tumours, in most cases, regress with glucocorticoid therapy and their histological differentiation from Leydig-cell tumors is quite difficult.
Objective
The aim of this study was to differentiate the histological and clinical features of the TARTs from those of the Leydig-cell tumours.
Methods
The authors report a case of bilateral Leydig-cell tumour associated with giant bilateral adrenal myelolipoma in a male with adrenogenital syndrome who was submitted to a bilateral orchiectomy.
Results
Testicular neoplasia continue to grow despite adequate hormonal treatment and a bilateral orchiectomy was performed. The histopathological examination of the specimen ultimately confirmed the diagnosis of bilateral Leydig-cell tumour.
Conclusions
This case shows the importance of all the relevant examinations, blood chemistry analysis, and instrumental tests in the differential diagnosis of TARTs and other testicular tumours.
Keywords
Introduction
Myelolipomas of the adrenal gland are nonfunctioning benign tumours, usually small in size and unilateral, composed of adipose cells and haematopoietic myeloid cells (1). They are usually asymptomatic, but rarely they can occur with pain, haemorrhage, necrosis and compression, in which case surgical exploration is required (2). Before the existence of newer imaging techniques, these tumours were discovered by chance at autopsy with an incidence of 0.08-0.2% (3). The frequency of detection is increasing with the advent of computed tomography (CT), magnetic resonance (MR) and ultrasonography. They are usually discoverded in late adult life with a mean age of presentation of 62 years, and men and women appear to be equally affected (4, 5). Myelolipoma is associated with excessive stimulation with adrenocorticotropic hormone (ACTH), and has been frequently reported in individuals with endocrine disorders such as adrenogenital syndrome, caused by 21α-hydroxylase deficiency (6).
Bilateral testicular masses may develop in patients with poorly controlled congenital adrenal hyperplasia (CAH) (7). In 1940, they were classified for the first time by Wilkins et al as testicular adrenal rest tumours (TARTs) (8). They have reported a prevalence of up to 94% in adult males with CAH and of 24% in male CAH children (9, 10). The etiology is not yet clear, but it would seem that a small number of adrenal cells migrate with gonadal cells with the descending testis because of their similar morphological features and because they both come from the coelomic epithelium (11). TARTs are not malignant and generally regress with corticosteroid therapy, but they resemble Leydig cell tumours and their histological differentiation is quite difficult. It is important to differentiate the particular features of TARTs in order to apply the appropriate treatment.
Leydig-cell tumours are rare testicular tumours of the male gonadal interstitium. They are frequently hormonally active, leading to feminizing or virilizing syndromes. The cause of this tumour is unknown and there are no known risk factors for developing this tumour. Leydig-cell tumours represent between 1% and 3% of adult testicular tumours, and 3% of testicular tumours in infants and children (12, 13). Peak incidence is in the third to sixth decade in adults, and between 3 and 9 years in children. Only 3% of cases are bilateral and about 10% are malignant tumours (12).
Below, we report an unusual presentation of bilateral Leydig-cell tumour associated with giant bilateral adrenal myelolipoma in a male with adrenogenital syndrome, caused by with 21α-hydroxylase deficiency.
Case Report
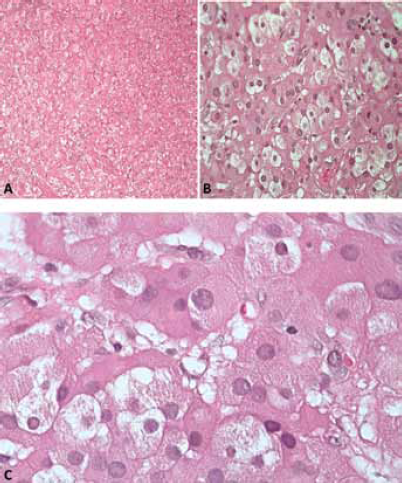
The patient, a 27-year-old male, was referred to our Urology Clinic in 1993, as he had two palpable bilateral testicular masses. A scrotal ultrasound examination revealed that they were hypoechogenic and hypervascular. He was found to be suffering from adrenogenital syndrome caused by a deficiency of 21α-hydroxylase, a condition that had been diagnosed when the patient was aged 4 and showed signs of precocious puberty. From that stage onwards, he was treated with replacement therapy of cortone acetate. His medical history indicated that he had been suffering from mental retardation and epilepsy, probably as a result of perinatal hypoxic injury. During his stay in hospital, a blood chemistry analysis showed that his levels of testosterone and luteinizing hormone (LH) were above normal and the follicle-stimulating hormone (FSH) was slightly increased, and an MR scan documented the absence of retroperitoneal lymphadenopathy and an increase of the left adrenal gland size (2.8 cm). It was considered advisable to subject the patient to an exploratory surgical procedure, given that during ultrasonography of the scrota, performed at regular intervals as requested by the endocrinologist in charge of the case, there were no signs of any reduction in the volume and dimensions in the testicular masses, despite years of replacement therapy. During the procedure, an immediate histological examination of a surgical specimen was carried out and showed the presence of a bilateral Leydig-cell tumour of the testis. Taking into consideration the fact that the patient was already azoospermic and complained of chronic bilateral pains, it was decided that we should proceed with radical surgery, and consequently, a bilateral orchiectomy without lymphadenectomy was performed. The subsequent histological analysis of the surgical specimens confirmed the initial diagnosis (Figs. 1, 2).
(
(
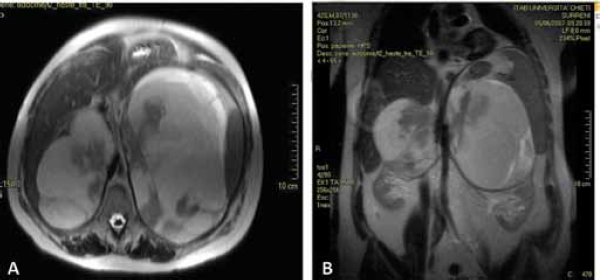
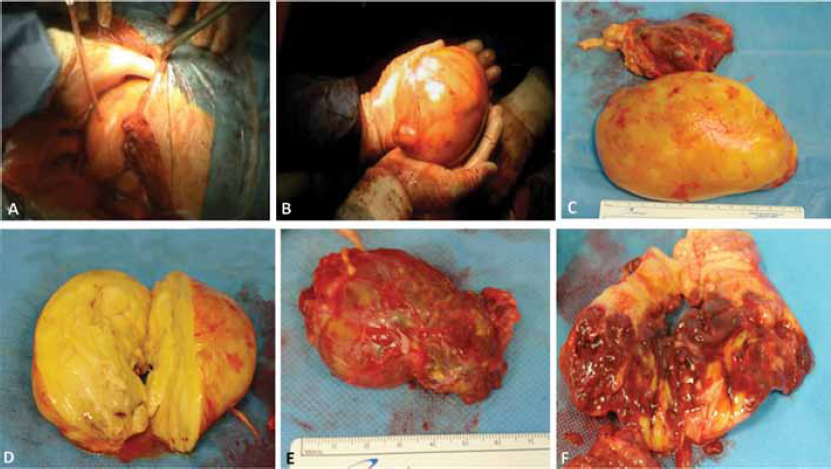
In 2003, the patient was already suffering from CAH because an MR and various ultrasound examinations of the abdomen revealed several bilateral adrenal lesions, hypervascularised, with dimensions of 11 cm on the right and 13 cm on the left. It was only 14 years later in December 2007 that the patient, now aged 42 and still receiving cortone acetate replacement therapy, was referred again to our Urology Clinic. He was complaining of recurrent abdominal pain, digestive problems and occasional vomiting. On MR examination, there emerged a detailed picture of two massive solid adrenal growths, one on the left side of the abdomen and the other on the right. Both were well-circumscribed with clearly defined margins and with maximum axial dimensions of 18 × 12 cm on the right side and 13 × 8 cm on the left side, respectively, and with a craniocaudal extension of about 17 cm on the left side and 11 cm on the right side (Fig. 3). From the haematological point of view, the tumour markers were within the norm. Given the severity of the symptoms, it was considered opportune to subject the patient to surgical intervention by performing a surrenalectomy and a resection of the solid masses, opting, however, for two separate procedures, as this was the patient's wish and equally so as not to subject the patient to excessive physical and psychological stress given his overall general clinical condition. Therefore, we restricted ourselves to remove the left-sided mass, as it was decidedly the larger. We gained access by a thoracic-freno-laparotomy method making an incision along the ninth intercostals space down to the median line. We did not manage to take away the mass in its entirety due to the difficulties encountered as a result of its massive size. However, we succeeded in removing the main growth as an integral part and the remainder was extracted in several pieces. The largest mass weighed 1200 g and had a maximum diameter of about 15 cm. The left adrenal gland had a diameter of about 7 cm (Fig. 4). An anatomopathological examination of the surgical specimen confirmed the diagnosis of adrenal myelolipoma with the growth being characterized by the presence of subatrophic residue from the adrenal gland (Fig. 5). During the period of postoperative convalescence, the patient was administered replacement therapy with Desametasone, and showed no complications or any electrolyte abnormalities, or symptoms relating to a deficiency of cortisol or mineralocorticoid hormones. From 2007 to the present day, the patient has received attentive follow-up with regular blood chemistry analysis and instrumental tests, and consultations with endocrinologists and urologists. In November 2010, the patient underwent a CT of the entire abdomen, without using any contrast, completed with dimensional reconstructions on the sagittal and coronal planes. In the right adrenal area, a massive formation was detected with maximum axial dimensions of about 18 × 12 cm, which was dishomogeneous due to the presence of solid adipose components, which were compatible with the adrenal myelolipoma, which displaces and compresses the liver and renal parenchyma (Fig. 6). Till the present day, the patient has not complained of any symptoms connected to the presence of this growth and has expressed a preference not to undergo any surgical intervention of removal of the right myelolipoma, and has expressed the wish to continue solely with pharmacological therapy. At the last blood chemistry analysis, the results were as follows: cortisol 6.9 µg/dl, testosterone less than 0.05 ng/ml (normal range 3-10), dehydroepiandrosteron-sulfate (DHEA-S) 2.3 µg/dl (normal range 96.9-391.5), Delta4-androstenedione 0.10 (normal range 0.30-2.63), ACTH 8.4 pg/ml (normal range 9.0-52.0), 17 alpha-hydroxyprogesterone 0.40 ng/ml (normal range 0.61-3.34) and erythrocyte sedimentation rate (ESR) 16 mm/h (normal range 1-12).
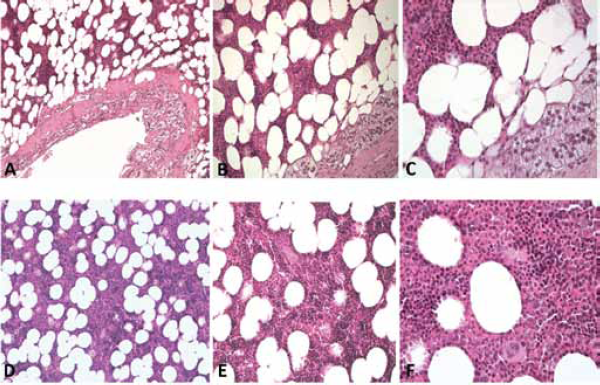
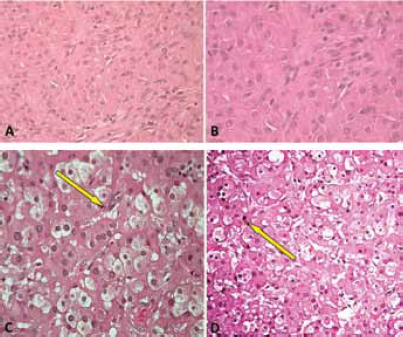
Histology of giant left adrenal myelolipoma.
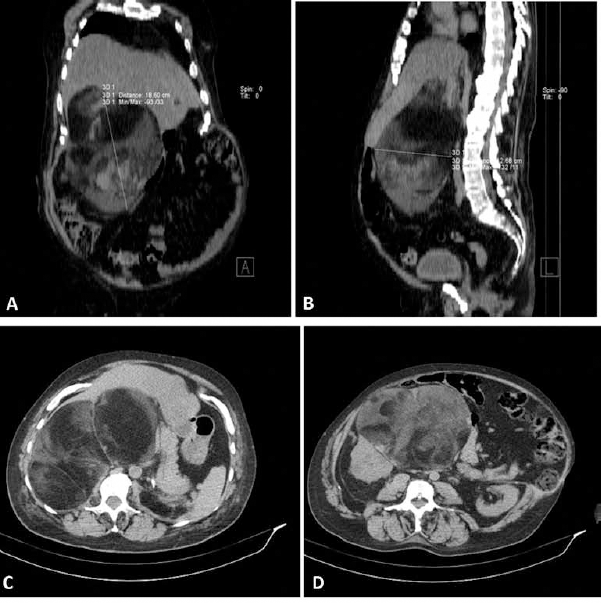
CT scan of abdomen showing expansive heterogenous tumour of fat-fluid
content, which displaces, compresses and does not infiltrate the adjacent
structures.
Discussion
Our case deals with a rare association between a giant bilateral myelolipoma tumour and a bilateral Leydig-cell tumour in a patient suffering from adrenogenital syndrome, caused by 21α-hydroxylase deficiency. In medical literature, no similar cases have been described when compared with the ever more frequent incidence of “adrenal rest” in such patients, and this has come about as a result of ever more accurate and precise imaging technology, which is now extensively accessible. In particular, scrotal echography, which is ever less costly and constantly improving in accuracy, has become of fundamental importance in the early diagnosis of such tumours. In 1995, Davis et al reported the case of a 37-year-old male patient with a rare association between adrenogenital syndrome and malignant Leydig-cell testicular tumour, with metastasis in the controlateral testicle and in the retroperitoneal lymph nodes. Despite surgical intervention of a bilateral orchiectomy and a retroperitoneal lymphadenectomy, after 6 months, the patient developed pulmonary metastasis (14).
The close histological similarity between TART and Leydig-cell tumour has been noted and described by numerous authors, but frequently determining a precise diagnosis that differentiates between the two has not proved straightforward. Leydig-cell tumours are bilateral in only 3% of cases and TART in more than 80% of cases (15). TARTs have vesicular nuclei without Reinke's crystals and a malignant degeneration has never been found in these tumours (16, 17).
Our patient had been receiving cortone acetate therapy since the age of 4, and from that point onwards, the results of his blood chemistry analysis have remained within normal limits with the exception of the FSH level, which in the preceding period was slightly above the normal level and led us to turn our attention to establishing a more accurate differential diagnosis. When he was 27, a testicular tumour was diagnosed that immediately gave rise to doubts regarding the initial diagnosis, as 23 years of replacement therapy should have considerably reduced the dimensions of the testicular neoplasia.
The fact that we were faced with bilaterally existing lesions prompted us to opt for a conservative surgical solution and an extemporaneous examination, despite the advanced age of the patient. In fact, such a technique is usually reserved for patients in whom an early diagnosis has been established, in order to safeguard residual healthy tissue and to prevent damage and azoospermia (18). The extemporaneous examination revealed that it was a Leydig-cell tumour, present bilaterally, and consequently a bilateral orchiectomy was carried out in accordance with international guidelines (19). The histopathological examination of the specimen ultimately confirmed the diagnosis with the following characteristics: yellow colour, vesicular nuclei with Reinke's crystals and absence of cordlike arrangement similar to the adrenal cortex (Fig. 2C, D). A lymphadenectomy was not necessary, as an MR of the abdomen carried out pre-operatively did not show any signs of any metastasis.
Conclusion
Our work, although dealing with a highly rare case, confirms the importance of careful follow-up examinations and observation when treating patients with adrenogenital syndrome, and also how imperative it is never to underestimate subsequent testicular masses, including bilateral masses, which can easily simulate TARTs that are more prevalent, but which could have a completely different prognosis.
Moreover, in the light of our results, we consider it vital to carry out all the relevant examinations, blood chemistry analysis and instrumental tests, so as to establish all the differences that might make a contribution in distinguishing between the two types of tumour, especially in cases wherein it is possible to establish an early diagnosis. The following are required: an objective and accurate examination, a testicular ecography and blood chemistry analysis for testosterone, FSH and LH.
Footnotes
Financial support: The authors have no financial disclosures to make.
Conflict of interest: The authors have no conflict of interest.