
Abstract
Epigenetic mechanisms are regulatory processes that control gene expression changes involved in multiple aspects of neuronal function, including central nervous system development, synaptic plasticity, and memory. Recent evidence indicates that dysregulation of epigenetic mechanisms occurs in several human epilepsy syndromes. Despite this discovery of a potential role for epigenetic mechanisms in epilepsy, few studies have fully explored their contribution to the process of epilepsy development known as epileptogenesis. The purpose of this article is to discuss recent findings suggesting that the process of epileptogenesis may alter the epigenetic landscape, affecting the gene expression patterns observed in epilepsy. Future studies focused on a better characterization of these aberrant epigenetic mechanisms hold the promise of revealing novel treatment options for the prevention and even the reversal of epilepsy.
The underlying cause of epilepsy in patients is still largely unknown, leaving 1 to 2% of the population without a cure and making the search for new treatments ever more urgent. Current treatment options for the epilepsies have been largely focused on and are effective at controlling seizure activity in most cases; however, cures for these disorders have been elusive. This is largely owing to the very complex progression of the disease state and the multiple inherited and acquired factors that can influence the onset and progression of epilepsy disorders. An emerging idea is that exploring epigenetic mechanisms, including covalent modifications of histones and DNA, may provide insight into how inherited or acquired alterations in the steady-state expression patterns of genes in the brain contribute to epileptogenesis.
During the past decade the science of epigenetics has advanced our understanding of the complex relationship between genetic and environmental factors that orchestrate expression of genes under numerous physiologic conditions. Accordingly, aberrant epigenetic regulation of genes has been implicated in several CNS disorders, including stroke, Alzheimer disease, schizophrenia, and depression (1, 2). Epilepsy is no exception, in that abnormalities in the expression and regulation of epigenetic factors offer a provocative locus for the integration of common pathways that may reveal novel insights into the development of this disease.
Brief Overview of Epigenetic Mechanisms in the CNS
Epigenetics has been defined as a set of self-perpetuating modifications on histone proteins or directly onto the DNA that produce alterations in chromatin structure as a direct consequence, and alterations in patterns of gene expression as an indirect consequence. This definition for epigenetics has long been associated with stable gene transcription regulation during normal neuronal development, cellular differentiation, and mitotic maintenance, but until recently was thought to be static in the brain following these processes. Epigenetic regulation has now been shown in the adult CNS to not only include persistent effects on gene expression patterns but to also dynamically coordinate gene transcription in response to environmental influences. Thus, epigenetic mechanisms have defining properties that make them well poised to act as regulators of chromatin structure in postmitotic neurons under both normal and disease states (1, 2).
The basic unit of chromatin is the nucleosome, and each nucleosome comprises 146 base pairs of double-stranded DNA wrapped around an octamer core of eight histone proteins (3, 4). Histones consist of a family of proteins that includes H1, H2a, H2b, H3, and H4, and their variants (i.e., H2a.X and H3.3). The H1 class of proteins is not a part of the nucleosomal core but is associated with the spacer DNA connecting the nucleosomal cores. The free N-terminal tails of histone proteins are unstructured and are amenable to addition or removal of functional groups that are defined as posttranslational histone modifications (PTM). These PTM serve to remodel the chromatin structure, thus allowing transcription factors (TF) and enhancer element binding proteins access to regulatory sites within DNA for the initiation of gene transcription.
To date, three PTM have been identified as dynamically regulated in the brain: acetylation, phosphorylation, and methylation (5). The addition of acetyl (i.e., H3K9,14) and phosphate (i.e., H3S10) functional groups to histone tails results in the exposure of the DNA and, hence, favors the formation of the transcriptionally active state of chromatin, or euchromatin. Unlike the charged acetyl and phosphate groups, the uncharged methyl groups on histone proteins are too small to disrupt the charge between histone proteins and associated DNA (chromatin) but instead regulate transcription by functioning as docking sites to recruit activator or repressor proteins to restructure chromatin (6). Furthermore, the addition of single or multiple methyl functional groups on histones can result in either the activation or repression of genes. For example, mono-, di-, and trimethylated forms of histone H3 at lysine 4 (H3K4me, H3K4me2, H3K4me3) and mono-methylation of histone H3 at lysine 9 (H3K9me) result in active gene transcription, whereas di- and trimethylation of histone H3 lysine 9 (H3K9me2, H3K9me3) result in the repression of gene transcription (7).
In addition to histones, DNA can also be epigenetically marked via methylation. Typically, cytosine nucleotides found within cytosine-guanine dinucleotide sequences linked by phosphodiester bonds (CpG sites) are methylated (8). DNA methylation is generally explicitly found in inactive gene promoter regions and associated with transcriptional gene silencing (9). However, investigations into the role of DNA methylation in the brain indicate that this epigenetic mechanism can facilitate both activation and repression of gene transcription in the CNS (7, 10). Furthermore, chromatin gene silencing via DNA methylation can be orchestrated by histone methylation markings that serve to recruit mac-romolecular complexes of epigenetic factors containing DNA-methylating enzymes. For example, the H3K9me2 and H3K9me3 marks can recruit DNA methyltransferases (DNMTs), a group of enzymes that catalyze DNA methylation, thus linking histone methylation to DNA methylation (11). In general, it has been observed that H3K4me3 negatively correlates with DNA methylation, whereas H3K9me2 and H3K9me3 positively correlate with DNA methylation (12). Thus, epigenetic mechanisms are not isolated events but rather they interact and influence each other.
Epigenetic mechanisms are complex, and the discovery of numerous proteins containing multiple catalytic and binding domains has created a large, confusing web of enzymes for the regulation of chromatin structure in a cell. In an effort to more clearly define the multiple mechanistic steps involved in gene expression via chromatin-based epigenetics, an operational model for these pathways has recently been proposed (13). This model is represented by three categories of signals that result in the alteration of the epigenetic state of a given gene. The first category involves an “Epigenator” extracellular signal, which comes from the environment and results in the activation of a second category of intracellular signals, or “Epigenetic Initiators,” that are necessary for the third category of “Epigenetic Maintainer” signals to sustain the chromatin environment. In this context, an Epigenetic Initiator can be any factor that would localize the chromatin complex assembly at a given gene.
To date, the identification of the key proteins and enzymes in each of these categories in the intact or diseased CNS is the subject of ongoing research. Of promise, recent studies have begun to shed light on how this exciting area of research relates to epileptogenesis, which is briefly discussed in this article. Tables 1 and 2 highlight examples of studies wherein aberrant regulation of epigenetic factors has been observed in human epilepsy disorders and in experimental epilepsy models. Additionally, the potential role of epigenetics in epileptogenesis is discussed in the context of alterations in molecular processes that can affect the development of epilepsy irrespective of the underlying cause, be it traumatic brain injury, other brain insults, or acquired genetic traits. Thus, epigenetic mechanisms can impact the acute deployment of genes resulting from seizures themselves or can have gradual effects on the steady-state expression profile of candidate genes that persist into epilepsy.
Examples of Studies in Which the Regulation of Epigenetic Mechanisms Has Been Observed With Human Epilepsy Syndromes
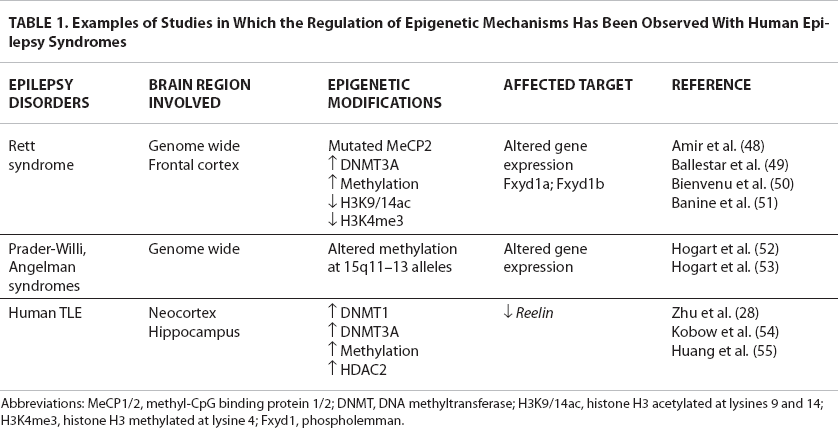
Abbreviations: MeCP1/2, methyl-CpG binding protein 1/2; DNMT, DNA methyltransferase; H3K9/14ac, histone H3 acetylated at lysines 9 and 14; H3K4me3, histone H3 methylated at lysine 4; Fxyd1, phospholemman.
Examples of Studies in Which the Regulation of Epigenetic Mechanisms Has Been Observed With Experimental Models of Epilepsy
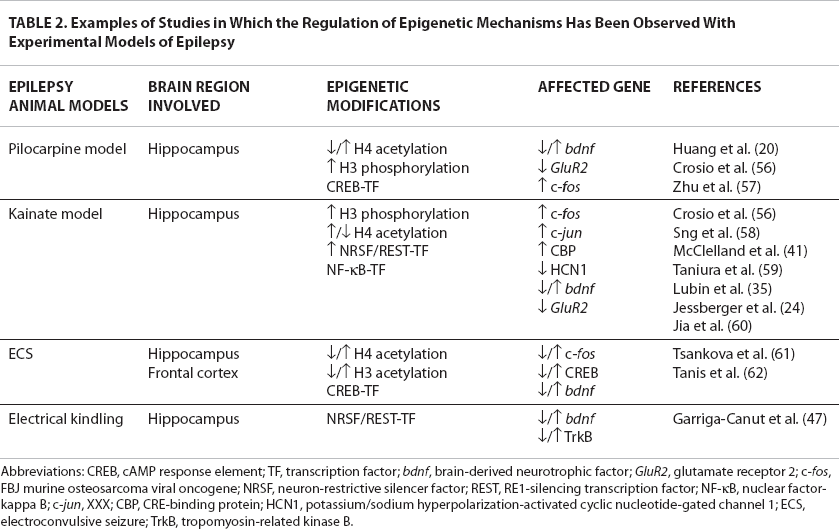
Abbreviations: CREB, cAMP response element; TF, transcription factor; bdnf, brain-derived neurotrophic factor; GluR2, glutamate receptor 2; c-fos, FBJ murine osteosarcoma viral oncogene; NRSF, neuron-restrictive silencer factor; REST, RE1-silencing transcription factor; NF-kB, nuclear factor-kappa B; c-jun, XXX; CBP, CRE-binding protein; HCN1, potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 1; ECS, electroconvulsive seizure; TrkB, tropomyosin-related kinase B.
Histone Acetylation in Epileptogenesis with Clinically Translational Applications
The addition of acetyl groups is carried out by a group of enzymes known as histone acetyl transferases (HAT) (14–19). A well-known example of a protein with HAT activity is the transcriptional co-activator p300 and cyclic adenosine monophosphate response element binding (CREB) protein-binding protein (p300/CBP), which interacts with the co-p300/CBP-associated factor (PCAF). Seizure activity results in gene expression changes, including alterations in mRNA levels for glutamate receptor 2 (GluR2) and brain-derived neurotrophic factor (bdnf), two well-characterized epileptogenesis-related genes. Of interest, HAT-mediated increases of histone acetylation levels at the promoter regions of the GluR2 and bdnf genes have been shown to correlate with their gene expression changes following seizures in an experimental animal model (20).
Histone deacetylases (HDAC) catalyze the removal of the acetyl group, and HDAC inhibitors (HDI) have a long history of use in the treatment of various neurologic disorders, including epilepsy. For example, valproic acid (VPA) has been used to control seizures for decades in patients with epilepsy (21) and has now been shown to be a potent inhibitor of class-I HDAC isoforms. Epileptogenic injuries are believed to induce postnatal neurogenesis in the dentate gyrus of the hippocampus, which may contribute to the development of epilepsy (22). Intriguingly, VPA has been shown to block neurogenesis and differentiation of hippocampal progenitor cells in vivo with corresponding increases in histone acetylation (23). In addition, VPA has been demonstrated to reverse cognitive impairments in a rodent model of epilepsy (24). VPA is believed to block the aberrant neurogenesis induced by seizure activity via its action as an HDI (24). Additional studies with other HDI demonstrate reversal of a seizure-induced decrease in histone acetylation at specific epileptogenesis-related genes like GluR2 (20). These pioneering studies suggest that, as the role of epigenetics in the etiology of epilepsy becomes clearer, additional characterization of more targeted HDI might reveal novel insights into the possible mechanisms and prevention of co-morbidities associated with epilepsy, such as aberrant neurogenesis and cognitive deficits.
The Potential Epileptogenic Role of DNA Methylation in Human Epilepsies is Undetermined
Extensive research on the role of DNA methylation in CNS development, cellular differentiation, behavior, and memory formation (6, 9) strongly supports the idea that DNA methylation may also play a crucial role in epileptogenesis. Methylated cytosines within DNA represent docking sites for proteins containing methyl-binding domains (25). Methyl-CpG binding protein-2 (MeCP2) recruits histone-modifying proteins that aid in the formation of the transcriptionally silent chromatin, or heterochromatin (25). MeCP2 is one of the first examples of a role for DNA methylation in the pathogenesis of epilepsy disorders (26). Additional evidence of DNA methylation in epilepsy include findings of hypermethylation of the reelin promoter in association with temporal lobe epilepsy (TLE) (27). In support of this idea, emerging evidence suggests that expression of DNMT enzymes, DNMT1 and 3a specifically, is increased in neurons from the temporal neocortex of TLE patients (28). Together, these findings suggest that additional studies are necessary to determine the function of DNA methylation in human epilepsies and experimental models of epileptogenesis.
Histone Methylation in Human Epilepsies with Indirect Evidence for a Role in Epileptogenesis
There is indirect evidence that histone methylation, like DNA methylation, may play a potential role in epileptogenesis. For example, JARID1C (also known as SMCX or KDM5C) is a histone demethylase that has been associated with X-linked mental retardation and in a significant minority population with epilepsy (12, 29–31). Intriguingly, investigations of dynamic histone methylation mechanisms in the CNS have recently been explored in synaptic plasticity and memory formation (7, 32). Such studies have revealed that histone methylation is critical to the process of memory formation (7). Thus, abnormal regulation of histone methylation marks in the brain can result in cognitive decline associated with human epilepsy syndromes; hence, these investigations should inspire future studies of the role that histone methylation plays in epileptogenesis and co-morbidities associated with epilepsy, such as cognitive deficits. In general, exploring histone methylation patterns in the epilepsies may help shed light on the molecular pathophysiology of these disorders.
TF Activation and Regulation of Epigenetic Mechanisms in Epileptogenesis
TF are important mediators of gene expression in the brain and are thought to play a major contributing role in epileptogenesis. Intriguingly, the cellular role of TF extends beyond simple binding to DNA to mediate gene transcription. Indeed, the binding of TF to DNA can result in the recruitment of epigenetically related co-activators or co-repressor complexes that ultimately influence chromatin restructuring. For example, the TF CREB (33) and nuclear factor-κB (NF-κB) have been shown to be activated following seizures (34, 35) and to associate with the p300 and CBP HAT proteins to remodel chromatin and further promote active gene transcription (36, 37). Other TF, such as the repressor element-1 silencing transcription factor/neuronal restrictive silencer factor (REST/NRSF) serve to repress gene expression through dynamic recruitment of epigenetic complexes (38, 39). Of interest, REST has been implicated in the regulation of several epileptogenesis specific factors, including growth factors, neurotransmitter receptors, and ion channels (40, 41). These examples illustrate the concept that chromatin structure and the modification of histone proteins can critically be influenced by TF, making them important candidates for study in epileptogenesis.
These newly identified roles for TF in the regulation of chromatin structure underscore the fact that there are several factors to consider in the context of epigenetic regulation of gene expression contributing to epileptogenesis. Thus, TF and the accessory proteins that they recruit allow for the balance between mRNA upregulation and downregulation, all in accordance with the epigenetic state of the genome. Future research efforts should focus on better characterizing the role of specific TF in the aberrant initiation of epigenetic factors in epileptogenesis. Such investigations may prove to be promising for the discovery and development of epileptic therapeutics.
Clinical Relevance
Because aberrant regulation of epigenetic mechanisms has been implicated in numerous human diseases, epigenetic-based drug therapy has been a hot topic of late. Here, we conclude with a discussion of the therapeutic potential of epigenetic treatments for the amelioration of epileptic conditions. An example of an epigenetic drug-treatment option for epilepsy and some of its co-morbidities is VPA. As previously discussed, VPA has been used for decades as an antiepileptic drug (AED) and recently was discovered to contain HDI properties (42, 43). However, these properties are non-specific, as VPA may also cause changes in the state of other epigenetic factors including methylation of genomic DNA (44). Thus, more targeted HDI, such as vorinostat (SAHA) and romidepsin, may prove to be therapeutically promising. Unfortunately, long-term treatment with these drugs is likely to be more harmful than helpful as VPA administration in pregnant animals and in pregnant epilepsy patients causes malformations or congenital birth defects (45, 46). Fortunately, HDI are only one class of currently FDA-approved epigenetic drugs. Another class includes the DNMT inhibitors (zebularine, 5-aza-cytidin), which have not been tested in the context of human epilepsy disorders or animal epilepsy models. In addition, the ketogenic diet is a treatment option for refractory epilepsy and may in part reduce seizures in experimental epilepsy animal models through the regulation of TF-like REST/NRSF (47). Finally, it remains to be determined whether other AED, like VPA, may exert their anticonvulsant properties specifically through the regulation of epigenetic mechanisms. Thus, additional studies are necessary to better clarify and understand the effect of AED on epigenetic mechanisms in epileptogenesis.
Conclusions and Future Direction
Epilepsies are very complex disorders, and an important fundamental issue that remains is how genetic and environmental conditions interact to contribute to their onset and progression. Indeed, much useful effort has been applied over the last decade to identify genes that influence epileptogenesis. However, in many instances it has been difficult to link specific genes with specific epilepsy syndromes. The emerging field of “epigenetics” studies biochemical processes, which by definition mediate interactions between environmental influences and the genome and trigger persisting effects on gene expression patterns in the cell. Thus, an exciting hypothesis to be further tested is that epigenetic processes regulate gene transcription changes in the nervous system during epileptogenesis, which may provide an alternative mechanism for epilepsy disorders that cannot be clearly linked to genes or environment alone. If this emerging hypothesis proves correct, progress in this area offers the potential to revolutionize our thinking about epileptogenic mechanisms and novel treatment strategies that involve manipulations of the epigenome.
Footnotes
Acknowledgements
I would like to thank Ryley Parrish and Rosemary Puckett for their thoughtful comments regarding the tables. This work was supported by the Epilepsy Foundation, McNulty Civitan International Scientist Award, and the Evelyn F. McKnight Brain Research Foundation.