
Abstract
Context
Sclerostin inhibits bone formation via the WNT/β-catenin pathway and is a therapeutic target in osteoporosis. Antibodies against sclerostin are currently under investigation for the treatment of osteogenesis imperfecta (OI), a rare, genetically and clinically heterogeneous bone fragility disorder. However, data on serum sclerostin levels in pediatric and adolescent patients with OI remain limited.
Design
This is a retrospective, cross-sectional analysis of serum sclerostin levels in a genetically heterogeneous cohort of children and adolescents with OI.
Patients and methods
80 serum samples from 74 OI patients (median age 8.9 years, range 0.1–20.7 years), classified by clinical severity and affected gene, were collected. Serum levels of sclerostin, osteoprotegerin (OPG), parathyroid hormone (PTH), alkaline phosphatase (AP) and 25-Hydroxy Vitamin D (25(OH)D) were measured and analyzed according to genotype and OI severity.
Results
The median serum sclerostin level in this cohort was 0.35 ng/mL (IQR 0.28–0.52). Disease severity showed an inverse correlation with serum sclerostin levels (Spearman ρ = −0.4547, 95% CI [−0.62, −0.25], P < 0.0001). Multivariable linear regression analysis revealed genotype-specific differences in sclerostin levels, particularly in patients with BMP1 or WNT1 mutations compared with other mutation subgroups. No significant correlations were found between sclerostin and OPG, PTH, 25(OH)D, or AP.
Conclusions
In this cohort of children and adolescents with OI, disease severity was inversely associated with serum sclerostin levels, and genotype-specific differences may reflect distinct pathophysiological mechanisms of bone metabolism. These findings may contribute identifying patient subgroups most likely to benefit from targeted anti-sclerostin therapies.
Introduction
Elucidating the different modifying pathways of bone formation and resorption led to new treatment approaches. Sclerostin, a glycoprotein encoded by SOST gene, is a potent inhibitor and negative modifier of bone formation through the WNT/β-catenin pathway. Secreted primarily by osteocytes, sclerostin suppresses osteoblast differentiation, thereby reducing bone formation. Moreover, it plays a role in the adaptation of the bone structure according to biomechanical needs.1–4 In murine models, overexpression of SOST has been associated with reduced bone mass, while genetic inactivation led to increased bone mass.5,6 Consistent with these findings, individuals with SOST loss-of-function mutations exhibit a high bone mass phenotype and excessive bone overgrowth.7,8 Although sclerostin is known to act primarily in a paracrine rather than an endocrine manner,9,10 circulating serum sclerostin levels have been shown to correlate with bone marrow plasma concentrations, suggesting that they reflect local bone metabolism. 11 Counterintuitively to findings on SOST expression, a positive correlation between serum sclerostin levels and bone mineral density (BMD) was observed and has been interpreted in different ways—either as a counter-regulatory mechanism within bone homeostasis or as an indirect reflection of the total number of osteocytes, under the assumption that this number is proportional to overall bone mass.12–14
Osteogenesis imperfecta (OI) is a rare genetic disease characterized by low bone mass, bone fragility, skeletal deformities and short stature. More than 90% of patients present with a mutation in one of the collagen genes COL1A1/A2 (collagen type I alpha 1 chain/collagen type I alpha 2 chain gene) leading to the altered bone structure. In the past decade, additional causative mutations have been identified in genes affecting diverse molecular pathways, including those influenced by sclerostin signaling.15,16
Based on its role in bone remodeling, sclerostin has emerged as a potential target of interest for therapeutic intervention in patients at risk of fractures. Recently, Romosozumab—an anti-sclerostin antibody—has been approved for the treatment of postmenopausal osteoporosis. 17 Furthermore, two phase 3 clinical trials are currently investigating anti-sclerostin antibodies (Romosozumab - NCT05972551, Setrusumab - NCT05768854) as potential treatments in children with OI. Preclinical studies in different murine OI models have shown promising results, including reduced fracture rates and increased cortical thickness and bone mass.18–21 These findings are further supported by the results of the phase 2b study of Setrusumab in adults with OI (NCT03118570), which demonstrated a dose-dependent increase in areal BMD of the lumbar spine and total radial volumetric BMD after 12 months of treatment. 22 Very recently, it was announced that NCT05768854 did not meet its primary endpoint of reducing the annualized clinical fracture rate, while improvements in bone mineral density were reported with strong statistical significance. 23
However, despite the growing interest in targeting sclerostin for OI treatment, data on circulating sclerostin levels in affected individuals—particularly children—remain scarce and inconsistent. In addition, previous studies that measured serum sclerostin levels in patients with OI24–28 primarily categorized patients according to clinical OI types (I–IV), without stratification based on the underlying causative mutations beyond COL1A1 or COL1A2. Consequently, potential genotype-specific differences in sclerostin regulation remain largely unexplored. Moreover, the interaction between sclerostin, osteoprotegerin (OPG) and RANKL (receptor activator of nuclear factor kappa-B ligand) has not been systematically investigated in pediatric patients with OI.
The aim of this study was to evaluate serum sclerostin levels in children and adolescents with different types of OI. We hypothesized that different OI genotypes may be associated with distinct sclerostin profiles.
Methods
Patients and sample collection
A total of 80 blood samples from 74 patients with OI were analyzed. All included patients were seen consecutively in our specialized clinic for skeletal dysplasia and had a genetically confirmed diagnosis of OI. Patients were clinically classified as having mild, moderate, or severe OI based on morbidity, mobility, and phenotypic presentation. Mildly affected patients were independent of assistive mobility devices and experienced fewer than two fractures per year. Moderately affected patients required assistive devices for daily mobility and had undergone surgical interventions to correct skeletal deformities. Severely affected patients were wheelchair-dependent and continued to experience fractures and skeletal deformities despite surgical intervention.
Genetic categorization was based on the affected gene, resulting in 10 distinct groups: COL1A1 (MIM 120,150), COL1A2 (MIM 120,160), CRTAP (MIM 605,497), LEPRE1 (MIM 610,339), FKBP10 (MIM 607,063), BMP1 (MIM 112,264), TAPT1 (MIM 612,758), SERPINF1 (MIM 172,860), PLOD2 (MIM 601,865), and WNT1 (MIM 164,820). Written informed consent for participation, data collection, analysis and publication was obtained from all patients and their legal guardians. Ethical approval was obtained from the local ethics committee of the University (approval number: 13-202).
Blood samples were collected during routine clinical visits and standard blood draws between August 2011 and August 2014. Two patients had multiple samples taken at different time points (one patient with five and another with three samples, respectively).
Laboratory measurements
Serum levels of sclerostin, OPG, parathyroid hormone (PTH), alkaline phosphatase (AP) and 25-Hydroxy Vitamin D (25(OH)D) were measured. PTH, AP and 25(OH)D levels were determined using standard laboratory methods.
Blood samples for sclerostin and OPG were collected in serum tubes, allowed to clot, and centrifuged to obtain serum. Serum was aliquoted and stored at −80°C until analysis. All samples were visually inspected prior to analysis, and samples with evidence of hemolysis were excluded. According to the manufacturer, both assays are stable under storage at ≤ −80°C for >24 months, and up to three freeze–thaw cycles do not affect analyte stability. The median storage duration of the analyzed samples was 7.0 months (IQR 5.7–11.1 months), and no sample exceeded 24 months of storage prior to measurement.
Sclerostin concentrations were measured using a high-sensitivity human Sclerostin ELISA kit (Ref. TE1023HS; TECOmedical Group). The assay has a limit of detection (LOD) of 0.008 ng/mL and a limit of quantification (LOQ) of 0.01–3.34 ng/mL, with a calibration range of 0–3 ng/mL. OPG levels were determined with the MicroVue OPG ELISA kit (Ref. 8034; QUIDEL Corporation), with a limit of detection of 0.13 pmol/L and a calibration range of 1.875–60 pmol/L All measurements were performed according to the manufacturers’ instructions.
Statistical analysis
All statistical analyses were performed using only the first measurement per patient (n = 74), thereby ensuring a patient-level unit of analysis. The total of 80 measurements is reported only for descriptive purposes.
Statistical analyses were conducted using GraphPad Prism version 10.3 and R version 4.6.0. Normality was assessed using the Shapiro-Wilk test. Since sclerostin levels were not normally distributed, data are presented as median (interquartile range, IQR). Serum sclerostin concentrations were additionally transformed into age- and sex-adjusted z-scores based on reference percentiles published by Fischer et al., 29 using the same assay kit. Median sclerostin concentrations and corresponding z-scores were analyzed using the Wilcoxon signed-rank test. Cohort-specific percentiles were used for descriptive visualization (Figure 3).
Group comparisons of sclerostin levels across disease severity groups (mild, moderate, severe) were performed using the Kruskal-Wallis test followed by pairwise Wilcoxon rank-sum (Mann-Whitney U) tests with FDR correction (Benjamini-Hochberg). Effect sizes were estimated as Hodges-Lehmann median differences with 95% confidence intervals.
Due to skewed distributions, both sclerostin concentrations and time since last antiresorptive treatment were log-transformed prior to regression analyses. Multivariable linear regression analyses were performed with log-transformed sclerostin concentrations as the dependent variable and age, sex, disease severity, mutation group, and log-transformed time since last antiresorptive treatment as independent variables. To account for multiple testing, false discovery rate (FDR) correction using the Benjamini-Hochberg procedure was applied and adjusted P-values (q-values) were used for statistical inference. A q-value <0.05 was considered statistically significant. Spearman correlation analyses were used to assess associations between sclerostin levels and disease severity, serum OPG, PTH, 25(OH)D and AP.
Results
Clinical data
Clinical data of the cohort.
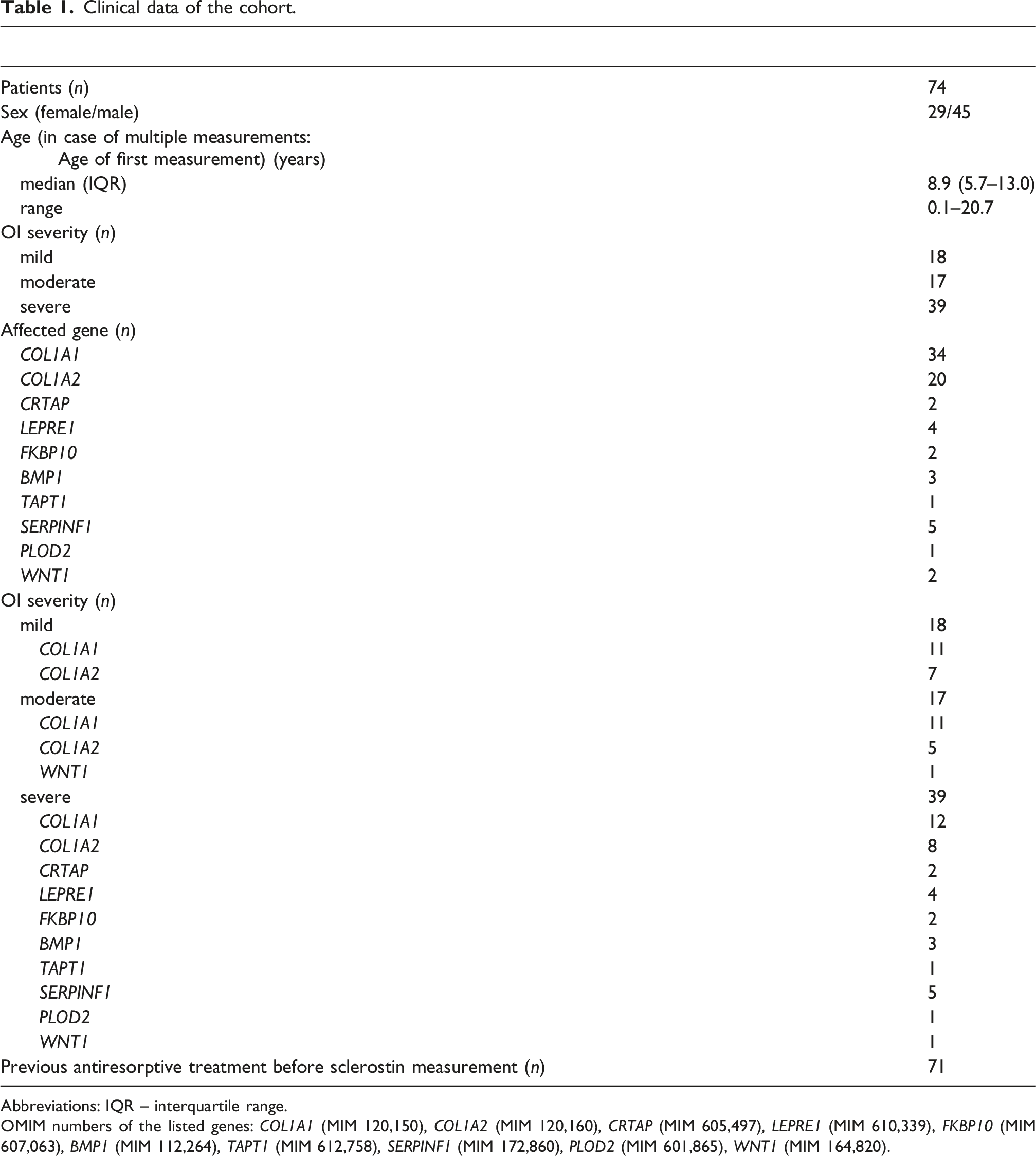
Abbreviations: IQR – interquartile range.
OMIM numbers of the listed genes: COL1A1 (MIM 120,150), COL1A2 (MIM 120,160), CRTAP (MIM 605,497), LEPRE1 (MIM 610,339), FKBP10 (MIM 607,063), BMP1 (MIM 112,264), TAPT1 (MIM 612,758), SERPINF1 (MIM 172,860), PLOD2 (MIM 601,865), WNT1 (MIM 164,820).
The most frequently affected genes were COL1A1 (n = 34) and COL1A2 (n = 20). Other causative genes included CRTAP (n = 2), LEPRE1 (n = 4), FKBP10 (n = 2), BMP1 (n = 3), TAPT1 (n = 1), SERPINF1 (n = 5), PLOD2 (n = 1), and WNT1 (n = 2). Of the 74 participants, 71 had received antiresorptive treatment (denosumab or bisphosphonates) prior to sclerostin measurement; for 70 patients, the exact time since the last bisphosphonate administration was available. Therapy duration until measurement and time since last dose for all patients, as well as for the two patients with multiple measurements, are provided in Tables S1 and S2.
Serum sclerostin levels
Sclerostin and OPG concentrations (median [IQR] or individual values).
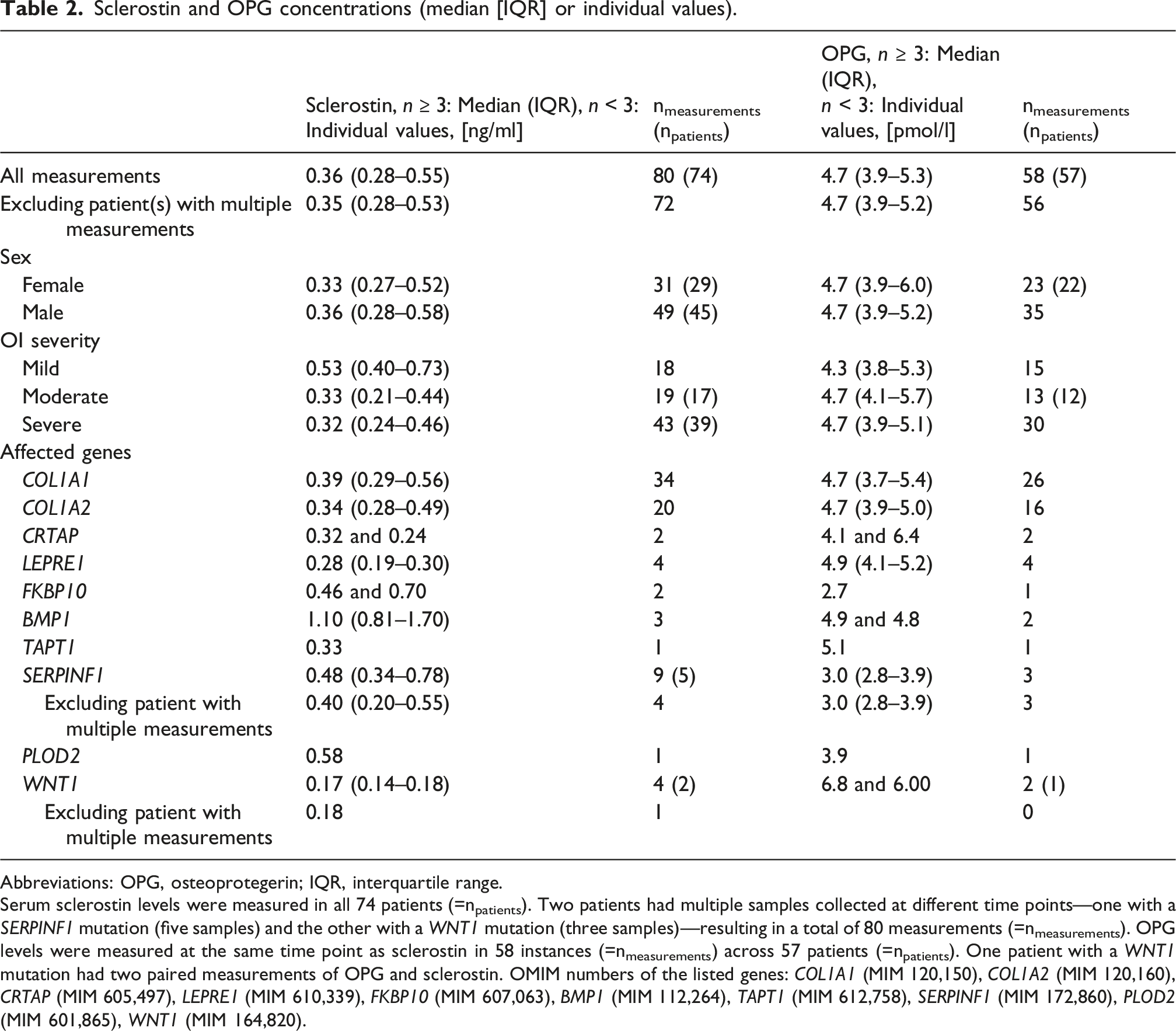
Abbreviations: OPG, osteoprotegerin; IQR, interquartile range.
Serum sclerostin levels were measured in all 74 patients (=npatients). Two patients had multiple samples collected at different time points—one with a SERPINF1 mutation (five samples) and the other with a WNT1 mutation (three samples)—resulting in a total of 80 measurements (=nmeasurements). OPG levels were measured at the same time point as sclerostin in 58 instances (=nmeasurements) across 57 patients (=npatients). One patient with a WNT1 mutation had two paired measurements of OPG and sclerostin. OMIM numbers of the listed genes: COL1A1 (MIM 120,150), COL1A2 (MIM 120,160), CRTAP (MIM 605,497), LEPRE1 (MIM 610,339), FKBP10 (MIM 607,063), BMP1 (MIM 112,264), TAPT1 (MIM 612,758), SERPINF1 (MIM 172,860), PLOD2 (MIM 601,865), WNT1 (MIM 164,820).
In this cohort, the median serum sclerostin level in children with OI (0.35 ng/mL [0.28–0.52], n = 74) was not significantly different from that observed in unaffected children (0.42 ng/mL [0.33–0.54], n = 424). Likewise, the median age-adjusted sclerostin z-score in this OI cohort (−0.33 [−0.94–0.58], n = 74) was not significantly different from the reference median of unaffected children (P = 0.0760). 29
Stratification by affected gene revealed varying sclerostin levels depending on the underlying mutation (Figure 1, Table 2). For example, in this cohort children with WNT1 mutations (n = 2) exhibited low sclerostin levels (below the 25th percentile of the overall cohort distribution), whereas children with BMP1 mutations (n = 3) showed elevated levels (above the 90th percentile of the overall cohort distribution).
30
Serum sclerostin concentrations (ng/ml) stratified by causative gene.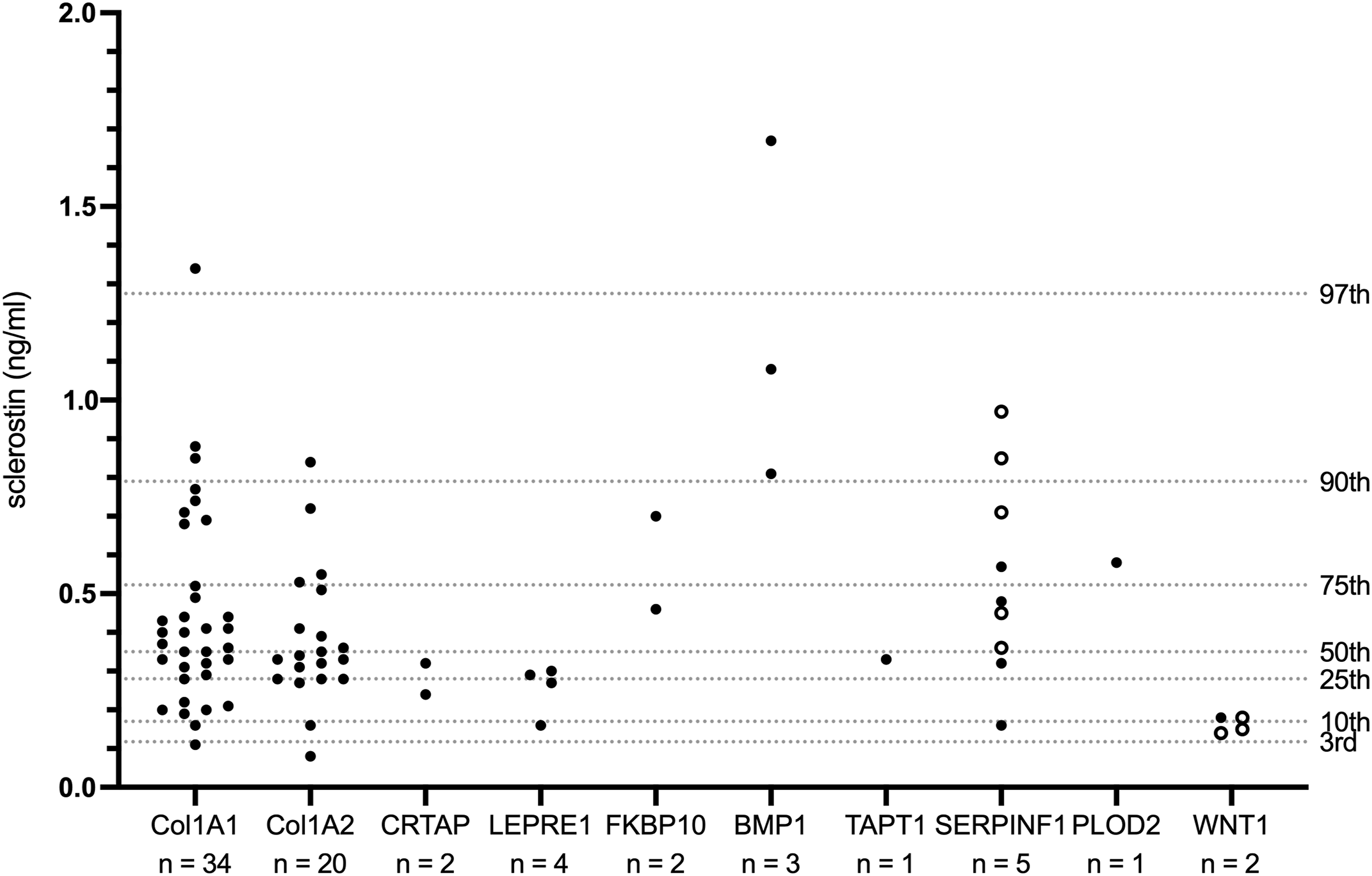
Within this cohort, increasing severity of OI was strongly associated with lower serum sclerostin levels (Spearman ρ = −0.4547, 95% CI [−0.62, −0.25], P < 0.0001, n = 74). Specifically, children with severe OI had significantly lower median sclerostin concentrations compared with those with mild OI (median [Hodges-Lehmann estimate] difference: −0.22 ng/mL, 95% CI [−0.35, −0.12], P < 0.0001, q = 0.0001). Similarly, moderately affected children showed significantly lower median sclerostin concentrations compared with those with mild OI (median [Hodges-Lehmann estimate] difference: −0.18 ng/mL, 95% CI [−0.33, −0.03], P = 0.005, q = 0.007), while no significant difference was observed between moderate and severe OI (Figure 2, Table 2). Serum sclerostin concentrations (ng/ml), stratified by OI severity. n = number of patients, q = FDR-adjusted P-values, ns = not significant.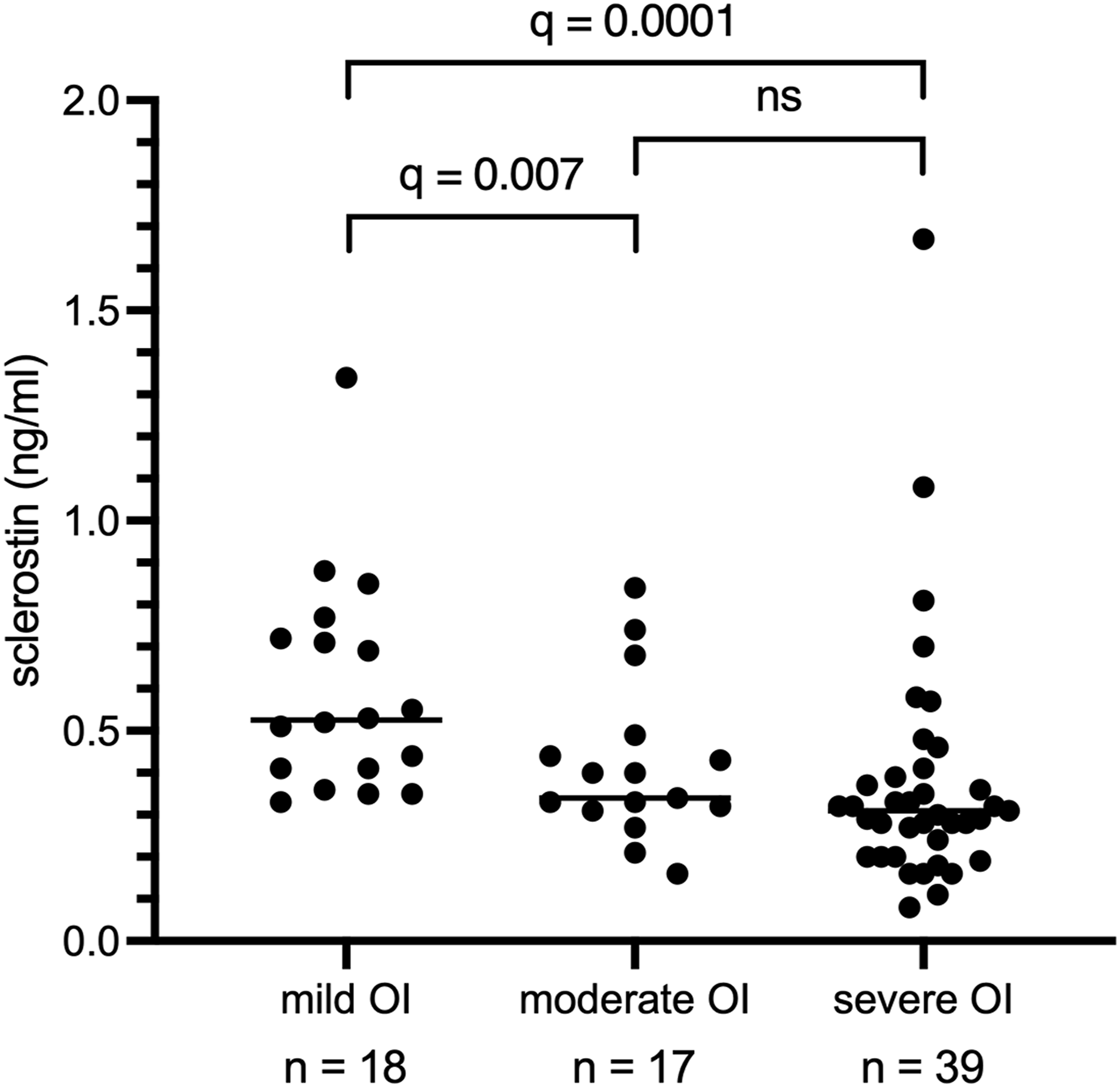
Multivariable linear regression analysis of serum sclerostin levels.
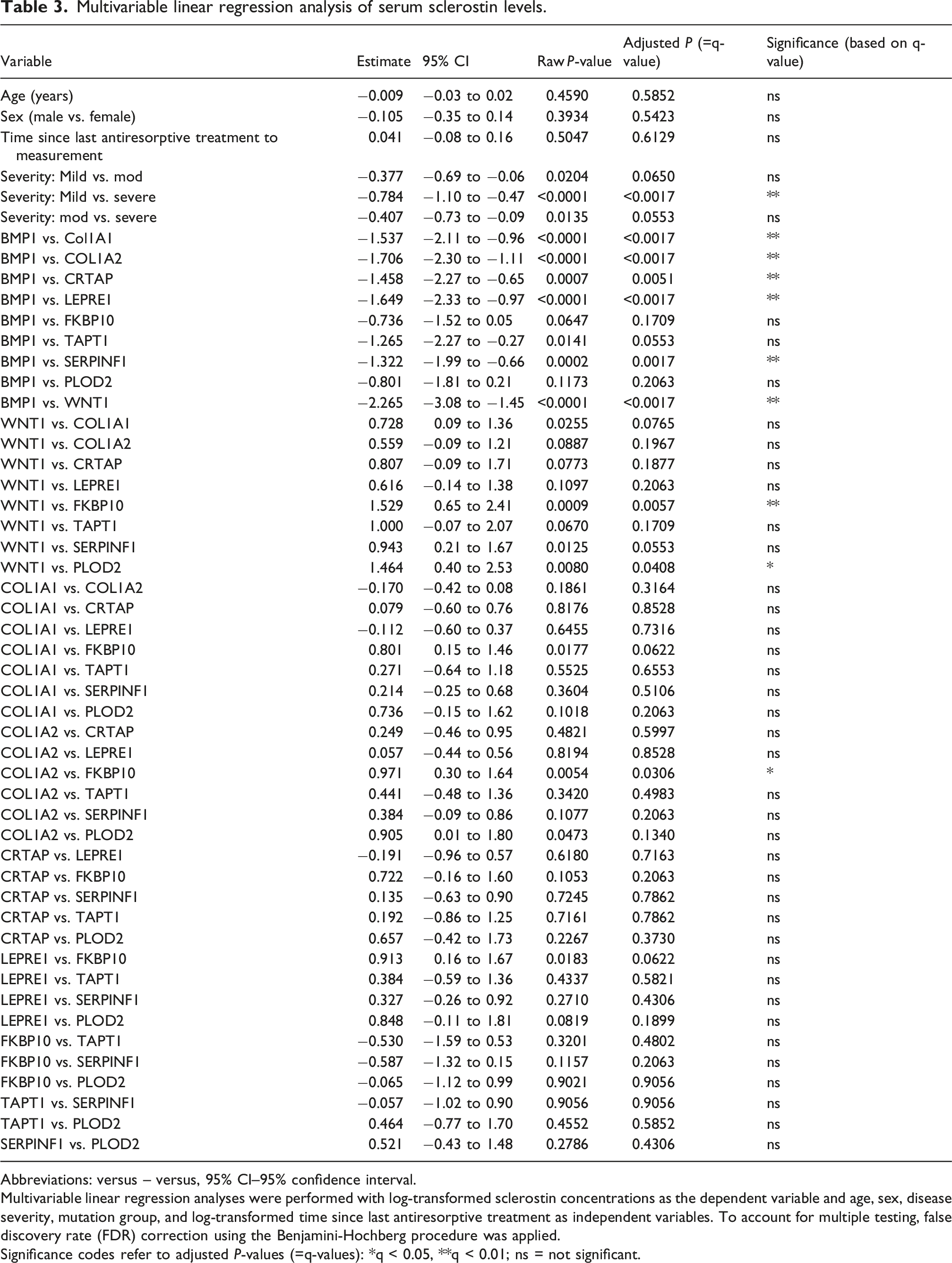
Abbreviations: versus – versus, 95% CI–95% confidence interval.
Multivariable linear regression analyses were performed with log-transformed sclerostin concentrations as the dependent variable and age, sex, disease severity, mutation group, and log-transformed time since last antiresorptive treatment as independent variables. To account for multiple testing, false discovery rate (FDR) correction using the Benjamini-Hochberg procedure was applied.
Significance codes refer to adjusted P-values (=q-values): *q < 0.05, **q < 0.01; ns = not significant.
Serum OPG levels
OPG levels were measured at the same time point as sclerostin in 58 instances across 57 patients (Figure 3(a), Table 2). One patient with a WNT1 mutation had two paired measurements of OPG and sclerostin. Serum OPG concentrations (pmol/l) (3A) and sclerostin/OPG ratio (3B) stratified by causative gene.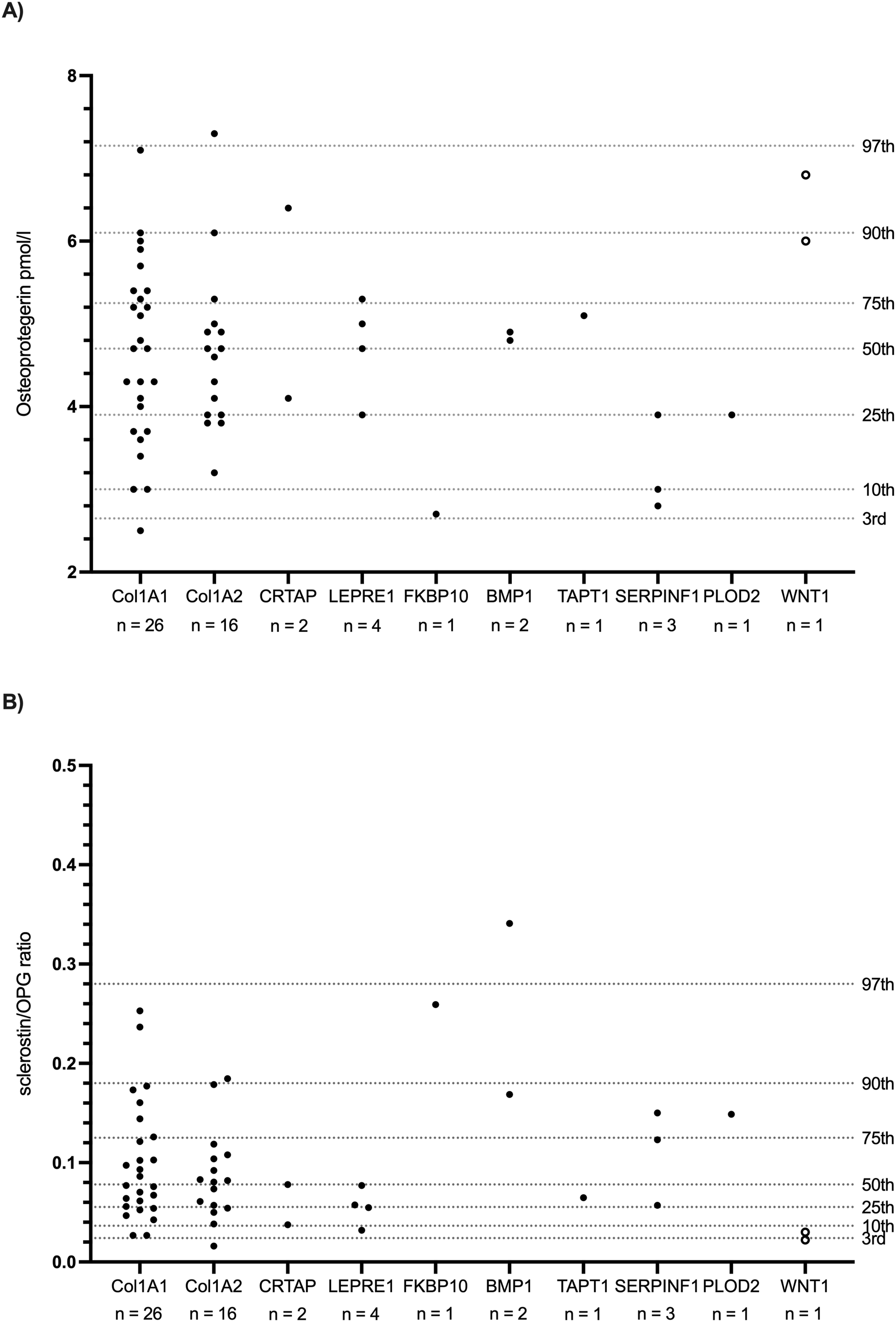
There was no correlation between serum sclerostin and OPG levels (Spearman ρ = −0.049, 95% CI [−0.31, 0.22], P = 0.7160, n = 57), although some mutation groups (FKBP10, PLOD2, WNT1) showed opposing trends with increased sclerostin-to-OPG ratios in FKBP10- and PLOD2-related OI and decreased ratios in WNT1-related OI (Figure 3(b)).
No correlation with PTH, 25(OH)D or AP
As observed for OPG, no significant correlations were observed between serum sclerostin levels and PTH, 25(OH)D or AP levels in this cohort.
Discussion
In this study, we evaluated serum sclerostin levels in children and adolescents with OI with regard to disease severity and genotype. To the best of our knowledge, this has not been previously investigated in a pediatric OI population.
The median serum sclerostin levels in this cohort were not significantly altered to those of unaffected children. Likewise, the median age-adjusted sclerostin z-score was not significantly different from the reference median of unaffected children. 29 Previous studies have reported conflicting results regarding serum sclerostin levels in patients with OI. Chen et al. 24 observed higher sclerostin concentrations in a mixed cohort of children and adults with OI, while studies in adult patients with OI (Nicol et al. 26 and Kocijan et al. 25 ) reported lower sclerostin concentrations. In contrast, our findings align with previous pediatric studies by Palomo et al. 27 and Brunetti et al., 28 which also reported no significant differences in serum sclerostin levels between children with OI and healthy controls. Notably, all of these studies categorized patients based on clinical OI type (I–IV), without differentiation based on causative mutations other than those affecting COL1A1 or COL1A2.
Considering the genetic heterogeneity of OI and the involvement of multiple molecular pathways, it is conceivable that sclerostin regulation varies depending on the specific mutation. Our findings support this notion: while most genotype-specific differences did not reach statistical significance in a multivariable regression model, likely due to small subgroup sizes, a significant difference in serum sclerostin levels was observed mainly in patients with BMP1 and WNT1 mutations in comparison to other mutation groups after adjustment for age, sex, disease severity and time since last antiresorptive treatment. Notably, patients with BMP1 mutations showed elevated sclerostin levels, while those with WNT1 mutations exhibited markedly low levels: BMP1 mutations impair procollagen processing and lead to increased collagen mineralization, resulting in a high bone mass OI phenotype. 34 The higher bone mass may reduce overall mechanical strains, which are key negative regulators of sclerostin expression by osteocytes.2,3 Consequently, reduced mechanical stimulation in BMP1-mutated patients may contribute to increased sclerostin production. In contrast, WNT1 mutations disrupt WNT/β-catenin signaling and suppress osteoblast-specific gene expression, resulting in impaired bone formation despite normal mineralization. 34 The markedly low sclerostin levels observed in these patients may reflect a compensatory mechanism to enhance osteoblast activity and promote bone formation. Our result is further supported by transcriptomic and proteomic data of patients with WNT1-related osteogenesis imperfecta which showed significantly downregulated expression of SOST compared to controls with COL1A1 mutation. 35 However, given the small genotype subgroup sizes (BMP1 n = 3 patients; WNT1 n = 2 patients), these findings, as well as their interpretation, should be considered exploratory. Additionally, as circulating sclerostin levels are also influenced by bone mass and osteocyte number, 36 the observed differences between BMP1 and WNT1 groups and other mutation groups may also be driven by individual variation in skeletal mass and osteocyte number rather than genotype-specific effects. Due to the retrospective nature of this study, there were no sufficient BMD data available to investigate this potential confounding factor.
We also found a significant inverse association between OI severity and serum sclerostin levels. Children with more severe forms of the disease had lower sclerostin concentrations compared to those with milder forms. This finding was stable after adjusting for age, sex, genotype and time since last antiresorptive treatment. While this may reflect a physiological adaptation aimed at enhancing bone formation in the context of severe skeletal fragility, as discussed above, it may also reflect differences in skeletal mass rather than a direct pathogenic mechanism. Two studies in adult patients with OI reported lower sclerostin levels in individuals with clinical OI type I compared to those with types III/IV, although the differences were not statistically significant.25,26 To the best of our knowledge, no other studies have specifically investigated the relationship between sclerostin levels and OI severity.
Regarding other bone markers, serum sclerostin levels did not correlate with OPG concentrations in this cohort. OPG, a glycoprotein secreted by osteoblasts and osteocytes, acts as a decoy receptor for RANKL, thereby inhibiting osteoclast differentiation and activity, and ultimately reducing bone resorption. 37 OPG is a WNT/β-catenin target gene and it has been shown that sclerostin, as an inhibitor of the WNT/β-catenin-pathway, suppresses OPG expression.38,39 In contrast to these expected mechanisms, a previous study in children with OI reported a positive correlation between serum sclerostin and OPG levels. 28 Interestingly, although no overall correlation between serum sclerostin and OPG levels was found in this cohort, mutation-specific trends emerged in our data. In selected subgroups, OPG levels showed opposing patterns to sclerostin concentrations, potentially reflecting distinct mutation-specific mechanisms of bone remodeling: The elevated OPG levels observed in patients with WNT1 mutation may seem surprising, considering that OPG is a target gene of WNT/β-catenin signaling. However, WNT1 is only one of several WNT ligands, 40 and there are also other factors such as estrogens that promote OPG gene expression independently of the WNT/β-catenin pathway.41–43 In the context of the concurrently observed low sclerostin levels—and thus reduced inhibition of the WNT/β-catenin pathway—along with the possibility that OPG expression can occur independently of WNT1, the elevated OPG levels likely represent a counter-regulatory mechanism to reduce osteoclast activity in WNT1-related OI. In contrast, patients with FKBP10- and PLOD2-related OI showed increased sclerostin-to-OPG ratios. Both mutations are known to impair collagen crosslinking, 34 leading to a structurally unstable extracellular matrix. Such matrix alterations may affect mechanotransduction in bone cells resulting in decreased OPG expression. 44 These mutation-specific changes may thus represent a compensatory response to increase bone stability.
Finally, we found no significant correlations between sclerostin levels and other bone metabolism markers, including PTH, 25(OH)D and AP. These findings appear to contrast with the known inverse regulation of SOST expression by PTH signaling, 45 as well as in studies in adults reporting negative correlations between serum sclerostin and circulating PTH levels.11,46–50 However, two studies in children—one in unaffected individuals, 31 and another in children with OI following bisphosphonate treatment 27 —similarly reported no correlation between sclerostin and PTH, supporting our findings in this cohort.
We also did not observe a correlation between serum sclerostin and 25(OH)D or AP. Reported associations in previous studies are inconsistent: some describe positive correlations in healthy children and in those with OI receiving bisphosphonate therapy, while others report no such correlation.27,31,51
In adults, a negative correlation between AP and sclerostin has been observed in elderly women but not in men. 52 These discrepancies may reflect age-related and/or sex-related differences as well as the influence of small sample sizes.
This study has several limitations. Due to the retrospective nature of the analysis, it was not possible to collect additional relevant clinical data, such as fracture rates or BMD—the latter known to be positively correlated with sclerostin levels. 51 Given rarity of OI, subgroup sizes—particularly when stratified by genotype—were small and the observed genotype-specific differences may therefore be individual-specific and related to clinical factors rather than disease biology. In particular, the BMP1 and WNT1 findings are based on very limited numbers of samples and should be interpreted with caution. Accordingly, these results should be considered exploratory. In nearly all cases, only a single measurement of serum sclerostin was available, limiting the assessment of intra-individual variability. This is particularly relevant given that sclerostin levels are regulated by multiple factors. 36 Another limitation is that, in the assay, only the standards and controls were performed in duplicates, but not the samples. Therefore, no intra- and inter-assay coefficients of variation could be calculated. However, all samples in the sclerostin assay and OPG assay respectively were run in one batch. Nonetheless, the study has notable strengths. Most importantly, it addresses a highly relevant clinical question in light of two ongoing phase 3 clinical trials investigating anti-sclerostin antibodies in children with OI (NCT05972551, NCT05768854). As these therapies may soon become part of the clinical care, baseline data on sclerostin variability across OI genotypes and severity levels are important. Our findings may contribute to identifying patients—based on their genotype—who are most likely to benefit from targeted anti-sclerostin treatment and may help guide future research into potential predictive markers of therapeutic response. This is of particular interest, as two recent studies in OI (including early results from NCT05768854) have shown that increases in bone mass do not necessarily correlate with a reduction in fracture rates, highlighting the need for genotype-based patient selection and individualized therapeutic strategies.23,53
Supplemental material
Supplemental material - Serum sclerostin levels in children with osteogenesis imperfecta
Supplemental material for Serum sclerostin levels in children with osteogenesis imperfecta by Susanna Reincke, Mirko Rehberg, Stefanie Stasek, Shino Junghänel-Welzing, Vera Fuessgen, Oliver Semler, and Heike Hoyer-Kuhn in Annals of Clinical Biochemistry
Footnotes
Acknowledgements
We sincerely thank all the patients and their families for their participation in the measurements, making this analysis possible. We also thank the DFG research unit FOR2722 (SE2373/1-2, Project: 384170921), which supported several of our authors (OS, MR, SS, HHK).
Consent to participate
Written informed consent was obtained from all patients and their legal guardians. No animal experiments were conducted in this study.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the “Ein Herz für Kinder” Foundation (MED-0054132).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Ethical approval was obtained from the local ethics committee of the University (approval number: 13-202).
Contributionship
SR wrote the first draft of the paper. OS and HHK designed the study. All data were collected during clinical visits with the support of MR, SJW and HHK. Data analysis was done by SR and revised by OS, SS and HHK. Revision and final approval of the manuscript were done by all authors. All authors agree to be accountable for the work and to ensure that any questions relating to the accuracy and integrity of the paper are investigated and properly resolved.
Guarantor
PD Dr. med. Heike Hoyer-Kuhn.
Supplemental material
Supplemental material for this article is available online.