
Abstract

Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a multi-organ syndrome resulting from defective regulation of inflammation in which excessive activation of macrophages and CD8+ lymphocytes causes a hyperinflammatory state mediated by high circulating levels of tumor necrosis factor–α (TNF-α), interferon-γ (IFN-γ), interleukin-10 (IL-10), IL-6, IL-8, IL-12, and IL-18.1,2 The resulting end-organ damage causes a severe, often fulminant systemic illness. HLH is thought to be either primary (familial), where there is a known genetic defect of lymphocyte function, or secondary in which case a trigger event may be identified, and there is no identified HLH causing mutation or family history of HLH. Since many of the clinical features seen in HLH are attributable to the underlying causative illness, HLH is often initially overlooked as a diagnosis. Minimizing delay in diagnosis is crucial for a favorable outcome. In this article, we outline some challenges that might be faced when evaluating patients for HLH and describe our experience managing nine pediatric patients with this disease.
Results/Clinical Experience
Table 1 presents the results and clinical experience of 9 patients diagnosed with HLH.
Nine Patients Diagnosed With HLH Based on HLH-2004 Diagnostic Criteria.
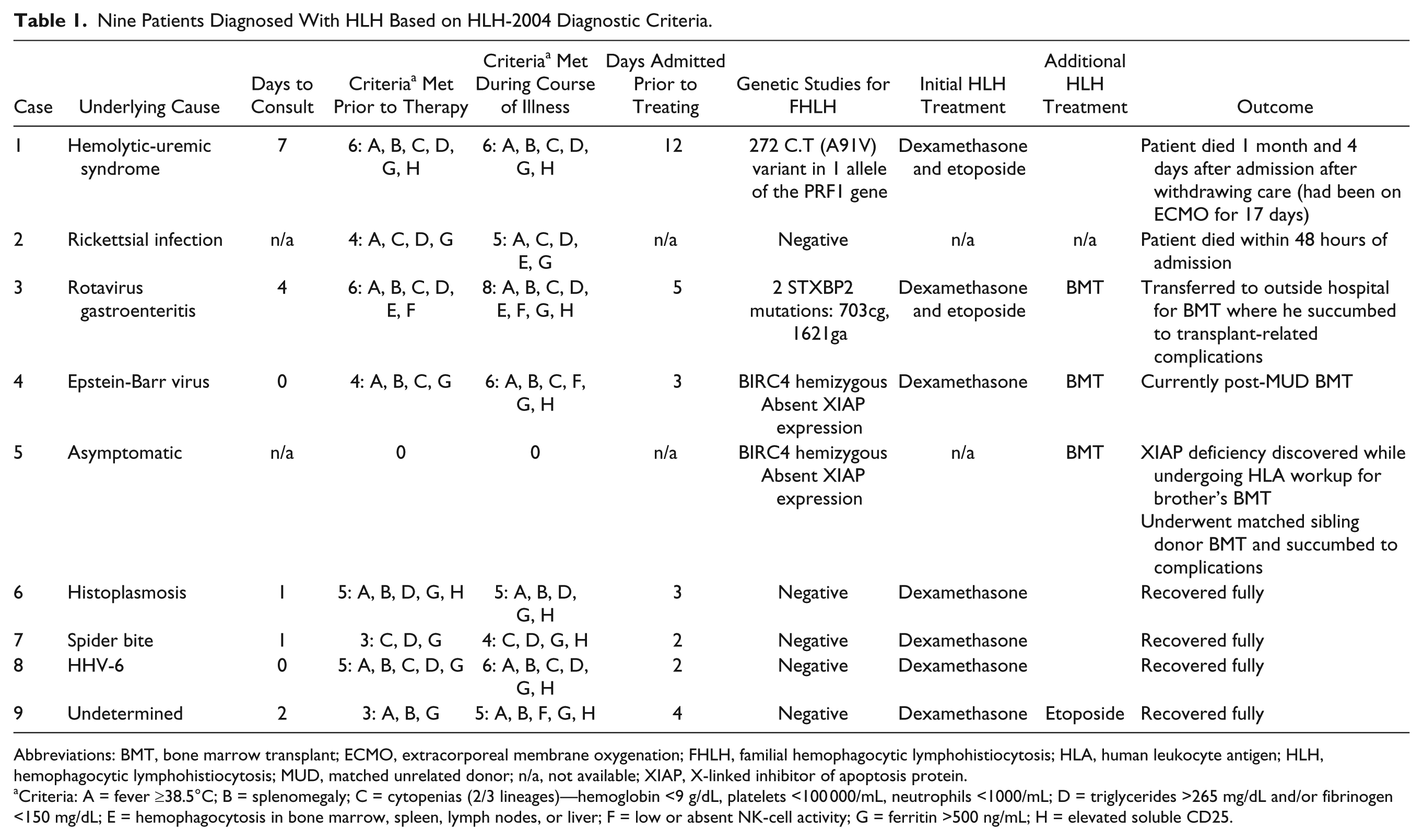
Abbreviations: BMT, bone marrow transplant; ECMO, extracorporeal membrane oxygenation; FHLH, familial hemophagocytic lymphohistiocytosis; HLA, human leukocyte antigen; HLH, hemophagocytic lymphohistiocytosis; MUD, matched unrelated donor; n/a, not available; XIAP, X-linked inhibitor of apoptosis protein.
Criteria: A = fever ≥38.5°C; B = splenomegaly; C = cytopenias (2/3 lineages)—hemoglobin <9 g/dL, platelets <100 000/mL, neutrophils <1000/mL; D = triglycerides >265 mg/dL and/or fibrinogen <150 mg/dL; E = hemophagocytosis in bone marrow, spleen, lymph nodes, or liver; F = low or absent NK-cell activity; G = ferritin >500 ng/mL; H = elevated soluble CD25.
Patient 1
A 5-year-old male presented with clinical features of atypical hemolytic-uremic-syndrome. HLH workup was commenced on day 10 of admission and HLH-directed treatment was commenced on day 12 (intrathecal hydrocortisone, dexamethasone, etoposide, and later alemtuzumab). Despite extracorporeal membrane oxygenation (ECMO) support, patient succumbed to his illness 1 month after admission.
Patient 2
A 5-year-old female presented with a 1-week history of fever, oliguria, melena, and petechial rash. She experienced rapid neurological deterioration and developed actue respiratory distress syndrome immediately after admission. Magnetic resonance imaging revealed profound anoxic brain injury and she died 48 hours after admission despite ECMO support before HLH-directed therapy could be initiated. Autopsy revealed hemophagocytosis in several organs and Rickettsial antigens in all tissues examined.
Patient 3
A 4-month-old male was admitted with high fever and gastroenteritis (rotavirus). On day 4 of admission, laboratory evidence of HLH was obtained and HLH-directed therapy was commenced on day 5 with dexamethasone and 50% dosing of etoposide. Soluble-CD25 was reported to be elevated on day 8 and natural killer (NK)–cell activity was reported as being absent on day 12. He harbored a known HLH-causing mutation and underwent a matched unrelated donor (MUD) cord blood transplant following HLH-2004 therapy. He ultimately succumbed to transplant-related complications.
Patient 4
A 16-year-old male with systemic Epstein-Barr virus syndrome met 4/8 HLH-2004 diagnostic criteria at admission. Dexamethasone/Rituximab were commenced. On day 13, soluble CD25 and NK-cell activity results confirmed the diagnosis of HLH. He was diagnosed with X-linked inhibitor of apoptosis protein (XIAP) deficiency and went on to receive a matched unrelated hematopoietic stem cell transplant. He is doing well 2 years post-transplant with no complications.
Patient 5
During pretransplant workup, the healthy 13-year-old brother of patient 4 was found to have XIAP deficiency. He underwent a matched sibling donor stem cell transplant but died from transplant-related complications.
Patients 6-9
Dexamethasone alone was commenced within 96 hours of admission. Three patients responded well to dexamethasone monotherapy; In patient 9, ferritin levels started to increase on day 6 of therapy so etoposide was added. None have had disease recurrence after completing HLH-2004-based therapy.
Discussion
While fever occurs in 91% of patients with HLH, it is ubiquitous in inflammatory, infectious, or rheumatological conditions and can be absent in neonates with HLH. 4 Splenomegaly, also the result of immune activation is commonly seen in systemic infections as well as rheumatological disorders. Splenomegaly may be missed during physical examination and may be absent in up to 16% of children with HLH. 3 Cytopenias in HLH are speculated to occur due to elevated levels of TNF-α and IFN-γ in the bone marrow microenvironment which suppress and/or cause apoptosis of blood cell precursors.5,6 Bone marrow suppression is also a common feature of many infectious, inflammatory and malignant disorders. Hypertriglyceridemia occurs in HLH because increased TNF-α and IFN-γ suppress the activity of lipoprotein lipase, 7 and hypofibrinogenemia is thought to occur because activated macrophages secrete plasminogen activator, leading to hyperfibrinolysis. 8 While both of these findings are fairly specific for HLH, hypertriglyceridemia is only seen in 60% to 70% of cases 9 and hypofibrinogenemia in 40%. 10 Hemophagocytosis resulting from macrophage activation in bone marrow, spleen, lymph nodes and/or liver is neither sensitive nor specific for HLH. 11 It is seen in approximately 50% to 60% of patients with HLH, 11 but can also be present following blood transfusions, in infection or autoimmune diseases in patients without HLH.12-14 NK-cell function assays are not widely available and may be difficult to interpret in profoundly leukopenic patients and patients receiving immunosuppressive therapy. There can be considerable variability in NK-cell function between individuals and in an individual over time. Concurrent NK-cell quantification testing should be performed as the test can be falsely positive if NK-cell number is reduced. Ferritin is released during cell breakdown 15 and at high levels can be specific for HLH. If HLH is suspected, blood samples should be diluted to accurately report high ferritin values (up to 100 000 ng/dL) so that the serum ferritin can be used to monitor disease response to treatment. Soluble CD25 (sCD25, the α-chain of the IL-2 receptor) reflects T-cell activation. Reference values vary significantly by age so the diagnostic criteria of >2400 units/mL is not applicable to all age groups. Being a send-out test for most institutions, time to result is typically 30 to 96 hours. Gender-based molecular diagnostic testing and mutation analysis for HLH is offered by referral centers. Perforin/granzyme B protein expression (which identifies PRF1 gene function), NK-cell surface CD107a expression (marker of exocytosis/degranulation), 16 Signaling lymphocyte activation molecule (SLAM)-associated protein expression (mutated in XLP1), and XIAP expression (mutated in XLP-2)16,17 serve as screening tests and have a relatively short turnaround time while definitive familial HLH (FHLH) genetic testing (AP3B1, LYST, SH2D1A, UNC13D, BLOC1S6, MAGT1, SLC7A7, CD27, PRF1, STX11, BIRC4, ITK, RAB27A, STXBP2) takes several weeks and does not affect acute patient management.
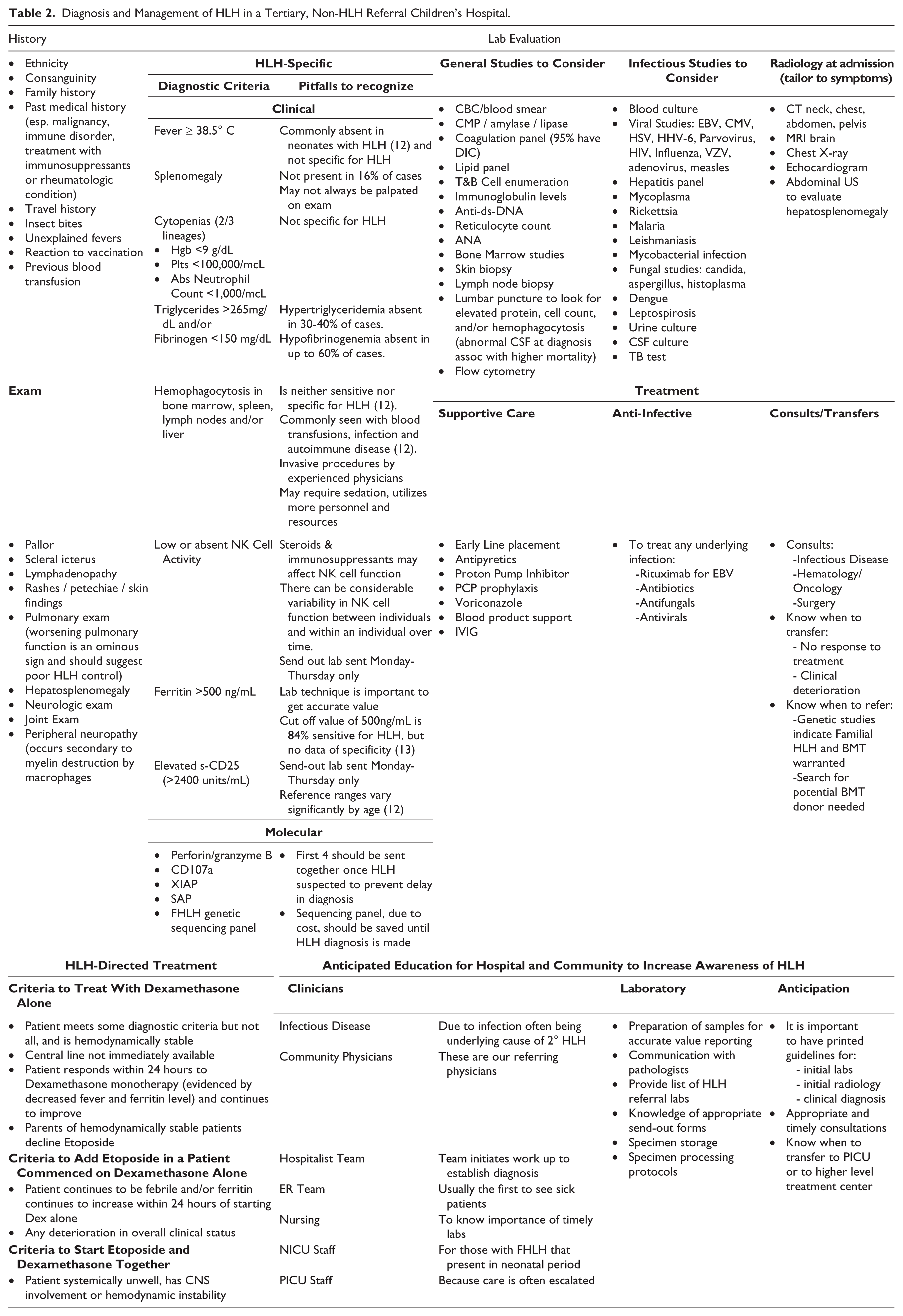
Hospital clinical services that could encounter patients with HLH should be trained to recognize HLH to reduce time to consultation. Early referral enables the pediatric hematology service to evaluate patients and coordinate laboratory evaluation to obtain all required samples prior to treatment and to have viable samples sent to outside laboratories early and to communicate with laboratories, send-out department, sedation services and HLH referral center if needed. Samples are best collected immediately prior to shipping in the evening and must be sent by overnight courier to arrive the following morning. Once a diagnosis of HLH is confirmed, FHLH studies can be performed which take about 4-6 weeks to result. In some patients who have not met all HLH diagnostic criteria, if the history and available clinical data are compelling, dexamethasone monotherapy can be commenced while awaiting results of the send-out tests and for central line placement. Once all diagnostic criteria are met, and depending on the patient’s clinical and laboratory response to dexamethasone monotherapy etoposide may be added to the treatment regimen (Table 2).
Diagnosis and Management of HLH in a Tertiary, Non-HLH Referral Children’s Hospital.
Author Contributions
LCM: concept, data collection, manuscript preparation, table preparation. KSF: concept, manuscript preparation, table preparation. RA: concept, manuscript preparation, table preparation.
Footnotes
Acknowledgements
The authors would like to thank Dr Rebecca Marsh for assistance in preparing this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.