
Abstract
Each month, subscribers to The Formulary Monograph Service receive 5 to 6 well-documented monographs on drugs that are newly released or are in late phase 3 trials. The monographs are targeted to Pharmacy & Therapeutics Committees. Subscribers also receive monthly 1-page summary monographs on agents that are useful for agendas and pharmacy/nursing in-services. A comprehensive target drug utilization evaluation/medication use evaluation (DUE/MUE) is also provided each month. With a subscription, the monographs are available online to subscribers. Monographs can be customized to meet the needs of a facility. Through the cooperation of The Formulary, Hospital Pharmacy publishes selected reviews in this column. For more information about The Formulary Monograph Service, contact Wolters Kluwer customer service at 866-397-3433.
Indications
Remdesivir is indicated for the treatment of coronavirus disease 2019 (COVID-19) requiring hospitalization in adults and pediatric patients 12 years and older and weighing at least 40 kg. Remdesivir should only be administered in a hospital or in a health care setting capable of providing acute care comparable to inpatient hospital care. 1
An emergency use authorization (EUA) for remdesivir has been issued by the food And Drug Administration (FDA) to allow for emergency use for the treatment of suspected or laboratory-confirmed COVID-19 in hospitalized pediatric patients weighing at least 3.5 kg.2,3 All requirements of the EUA must be met prior to administration. 2
Clinical Pharmacology
Remdesivir is a nucleotide analog inhibitor of viral RNA-dependent RNA polymerase, with activity against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), Middle East respiratory syndrome (MERS-CoV), and a variety of other coronaviruses, as well as other RNA virus families including paramyxoviruses, pneumoviruses, and filoviruses.1,4-6 Remdesivir is an adenosine nucleotide prodrug that distributes into cells where it is metabolized to a nucleoside monophosphate intermediate by carboxyesterase 1 and/or cathepsin A, depending on the cell type. The nucleoside monophosphate is subsequently phosphorylated by cellular kinases to form a pharmacologically active nucleoside triphosphate metabolite. Remdesivir triphosphate acts as an analog of adenosine triphosphate (ATP) and competes with high selectivity (3.65-fold) over the natural ATP substrate for incorporation into nascent RNA chains by the SARS-CoV-2 RNA-dependent RNA polymerase. Incorporation into the RNA chain results in delayed chain termination (position i+3) during replication of the viral RNA. When remdesivir nucleotide is present in the viral RNA template, the efficiency of incorporation of the complementary natural nucleotide is compromised, inhibiting viral RNA synthesis. Remdesivir triphosphate is a weak inhibitor of mammalian DNA and RNA polymerases, including human mitochondrial RNA polymerase. 1
Remdesivir exhibited antiviral activity against a clinical isolate of SARS-CoV-2 in cultured primary human airway epithelial cells, with a 50% effective concentration (EC50) of 9.9 nM after 48 hours of treatment. It inhibited replication of SARS-CoV-2 in the continuous human lung epithelial cell line Calu-3, with an EC50 value of 280 nM after 72 hours of treatment. 1 Additional clinical trial data are being assessed to determine the impact of remdesivir on viral shedding and viral load. 7
No clinical or cell culture data are available on the development of SARS-CoV-2 resistance to remdesivir, although introduction of F480L and V557L substitutions into SARS-CoV resulted in a 6-fold reduction in susceptibility to remdesivir in cell culture. 1 An additional study is being conducted to identify remdesivir-resistant SARS-CoV-2 variants and characterize several independent isolates phenotypically and genotypically. 7
In an animal model, rhesus macaques treated with remdesivir early in infection did not show signs of respiratory disease, unlike vehicle control animals; macaques treated with remdesivir also showed reduced pulmonary infiltrates on radiographs and lower lung viral loads and reduced lung damage on necropsy. 8
Pharmacokinetics
Peak concentrations of remdesivir are reached within a median of 0.67 to 0.68 hours following a 30-minute intravenous (IV) infusion. Remdesivir plasma concentrations decline rapidly and are accompanied by appearance of the GS-704277 metabolite (peaks in 0.75 hours) and the nucleoside GS-441524 metabolite (peaks within 1.51-2 hours).1,9 The GS-441524 metabolite is the predominant plasma metabolite. 9
Remdesivir is 88% to 93.6% bound to plasma proteins, while the metabolites are minimally plasma protein bound (1%-2%). 1
The median plasma elimination half-life of remdesivir is 1 hour; median half-life of the GS-441524 metabolite is 27 hours, and that of the GS-704277 metabolite is 1.3 hours.1,9 Remdesivir is metabolized via CES1 (80%), cathepsin A (10%), and CYP3A (10%) to 2 major metabolites. The GS-704277 metabolite undergoes further metabolism via HINT1, while the GS-441524 metabolite is not significantly metabolized but is primarily eliminated via glomerular filtration and active tubular secretion. The percentage of dose excreted in the urine is 10% for remdesivir, compared with 49% for the GS-441524 metabolite, and 2.9% for the GS-704277 metabolite. 1
Pharmacokinetic differences in remdesivir exposure based on sex, race, age, renal function, and hepatic function have not been assessed. The pharmacokinetics of remdesivir have not been evaluated in pediatric patients or in patients with renal or hepatic impairment. 1 Based on modeling and simulation, the recommended dosing regimen for patients 12 years and older and weighing at least 40 kg is expected to produce comparable steady-state plasma exposures of remdesivir and metabolites as observed in healthy adults. 1 Studies will be conducted to identify remdesivir drug interactions and effects on the QTc interval, as well as to assess remdesivir pharmacokinetics and safety in pediatric patients; pregnant patients; subjects with moderate and severe hepatic impairment; and subjects with mild, moderate, and severe renal impairment. 7
Comparative Efficacy
Indication: Coronavirus Disease 2019
Guidelines
Studies
● Time to recovery (defined as the first day during the 28 days after enrollment on which a patient met criteria for category 1, 2, or 3 on the 8-category ordinal scale) was shorter in the remdesivir group than in the placebo group (median, 10 days vs 15 days; rate ratio for recovery, 1.29 [95% CI, 1.12-1.49]; P < .001). Among the 957 patients with severe disease at enrollment, median time to recovery was 11 days with remdesivir compared with 18 days with placebo (rate ratio for recovery, 1.31; 95% CI, 1.12-1.52).
● Clinical status at day 15, as assessed on the 8-category ordinal scale, was more likely to be improved in the remdesivir group than in the placebo group (odds ratio [OR] for improvement, 1.5 [95% CI, 1.2-1.9]). ● Time to improvement of 1 category on ordinal scale was shorter in the remdesivir group (median, 7 days vs 9 days; rate ratio for recovery, 1.23 [95% CI, 1.08-1.42]). ● Time to improvement of 2 categories on ordinal scale was shorter in the remdesivir group (median, 11 days vs 14 days; rate ratio for recovery, 1.29 [95% CI, 1.12-1.48]). ● Time to discharge or a National Early Warning Score of 2 or less maintained for 24 hours, whichever occurred first, was shorter in the remdesivir group (median, 8 days vs 12 days; rate ratio, 1.27 [95% CI, 1.1-1.46]). ● Median number of days with supplemental oxygen among the 913 subjects receiving supplemental oxygen at enrollment, was lower in the remdesivir group (median, 13 days vs 21 days; difference, −8 days [95% CI, −11.8 to −4.2]). Percentage of patients not receiving oxygen at enrollment and requiring new use of oxygen did not differ (36% in the remdesivir group vs 44% in the placebo group; difference, −8 days [95% CI, −24 to 8]). ● Median number of days of noninvasive ventilation or high-flow oxygen use in the 193 patients receiving these interventions at baseline was 6 in both groups. ● Incidence of new noninvasive ventilation or high-flow oxygen use among the 573 patients not receiving noninvasive ventilation or high-flow oxygen at baseline was lower in the remdesivir group (17% vs 24%; difference, −7% [95% CI, −14% to −1%]). ● Median number of days of mechanical ventilation or ECMO during study among the 285 patients receiving these interventions at baseline was 17 in the remdesivir group and 20 in the placebo group (difference, −3 days; 95% CI, −9.3 to 3.3). ● Percentage of the 766 patients not receiving invasive ventilation or ECMO at enrollment and requiring new use of these interventions during the study was lower in the remdesivir group (13% vs 23%; difference, −10% [95% CI, −15% to −4%]). ● Median duration of initial hospitalization (up to day 29) was 12 days in the remdesivir group and 17 days in the placebo group (difference, −5 days; 95% CI, −7.7 to −2.3); readmission occurred in 5% of patients in the remdesivir group and in 3% in the placebo group (difference, 2%; 95% CI, 0% to 4%). ● Kaplan-Meier estimates of mortality by day 15 were 6.7% in the remdesivir group and 11.9% in the placebo group (HR, 0.55 [95% CI, 0.36-0.83]). ● Kaplan-Meier estimates of mortality by day 29 were 11.4% in the remdesivir group and 15.2% in the placebo group (HR, 0.73 [95% CI, 0.52-1.03]).
● Time to clinical improvement (defined as a 2-point reduction from admission in clinical status on the 6-point ordinal scale or hospital discharge, whichever came first) within 28 days after randomization did not differ between the remdesivir and placebo groups in the intention-to-treat (ITT) analysis population (median, 21 days in the remdesivir group compared with 23 days in the placebo group; HR, 1.23 [95% CI, 0.87-1.75]). Results were similar in the per-protocol analysis (21 days vs 23 days; HR, 1.27 [95% CI, 0.89-1.8]). Among subjects treated within 10 days of symptom onset in the ITT population, median time to improvement was 18 days in the remdesivir group and 23 days in the placebo group (HR, 1.52; 95% CI, 0.95-2.43); however, the difference was not statistically significant.
Proportion of patients in each clinical category did not differ significantly between treatment groups at day 7 (OR, 0.69; 95% CI, 0.41-1.17), day 14 (OR, 1.25; 95% CI, 0.76-2.04), or day 28 (OR, 1.15; 95% CI, 0.67-1.96). All-cause mortality at day 28 was 14% in the remdesivir group and 13% in the placebo group (difference, 1.1%; 95% CI, −8.1% to 10.3%). ● Duration of invasive mechanical ventilation was 7 days in the remdesivir group and 15.5 days in the placebo group (difference, −4 days; 95% CI, −14 to 2). Duration of oxygen support did not differ between groups (19 days with remdesivir and 21 days with placebo; difference, −2 days [95% CI, −6 to 1]). Duration of hospital stay did not differ between groups (25 days with remdesivir and 24 days with placebo; difference, 0 days [95% CI, −4 to 4]).
● Viral loads were similar to baseline and declined over time to a similar extent in both groups.
● Clinical improvement of at least 2 points on the 7-point ordinal scale at day 14 occurred in 64% of patients treated with the 5-day course and in 54% of those treated with the 10-day course. After adjustment for the imbalance in baseline clinical status, patients treated with the 10-day course had a distribution in clinical status at day 14 similar to that in patients treated with the 5-day course (P = .14).
● Proportion of patients with adverse events that occurred on or after the first dose of remdesivir for up to 30 days after the last dose was similar in the 2 groups (70% in the 5-day group and 74% in the 10-day group).
● Time to clinical improvement (defined as an improvement of at least 2 points from baseline on the 7-point ordinal scale) was 10 days in the 5-day group and 11 days in the 10-day group (difference, 0.79; 95% CI, 0.61-1.01). ● Proportion of patients achieving recovery (defined as an improvement from a baseline ordinal scale score of 2 to 5 to a score of 6 or 7) was 64% in the 5-day group and 54% in the 10-day group (baseline-adjusted difference, −6.3%; 95% CI, −15.4% to 2.8%). Median time to recovery was 10 days in the 5-day group and 11 days in the 10-day group (difference, 0.81 days; 95% CI, 0.64-1.04). ● Proportion of patients achieving modified recovery (defined as improvement from a baseline ordinal scale score of 2 to 4 to a score of 5 to 7, or from a score of 5 to a score of 6 or 7) was 70% in the 5-day group and 59% in the 10-day group (difference, −6.7%; 95% CI, −15.3% to 1.9%). ● Median time to modified recovery was 9 days in the 5-day group and 10 days in the 10-day group (difference, 0.82 days; 95% CI, 0.64-1.04). ● By day 14, 8% of patients in the 5-day group and 11% in the 10-day group had died. ● Among patients receiving mechanical ventilation or ECMO at day 5, 40% in the 5-day group died by day 14 compared with 17% in the 10-day group. Improved outcomes with treatment beyond 5 days were not observed in patients receiving noninvasive positive-pressure ventilation or high-flow oxygen, in patients receiving low-flow oxygen, or in patients breathing ambient air.
Results cannot be applied to patients receiving mechanical ventilation; further evaluation of this subgroup, and of other high-risk groups such as immunocompromised persons, is needed to determine the shortest effective duration of therapy.
Clinical status on day 11 (as assessed on 7-point ordinal scale) was better in the 5-day remdesivir group than in the standard care group (OR, 1.65 [95% CI, 1.09-2.48]; P = .02), but did not differ significantly between the 10-day remdesivir group and the standard care group (P = .18).
● Proportion of patients experiencing adverse events was 51% in the 5-day group, 59% in the 10-day group, and 47% in the standard care group. The difference between the 10-day group and standard care group was significant (difference, 12% [95% CI, 1.6%-21.8%]; P = .02).
● No differences were observed between the 5-day and 10-day remdesivir groups and the standard care group for any of the following exploratory end points: time to 2-point or greater improvement in clinical status, time to 1-point or greater improvement in clinical status, time to recovery, time to modified recovery, and time to discontinuation of oxygen support. ● Kaplan-Meier estimates of all-cause mortality at 28 days were 1% (95% CI, 0%-2.6%) for the 5-day group (P = .43 vs standard care), 2% (95% CI, 0% to 3.6%) for the 10-day group (P = .72 vs standard care), and 2% (95% CI, 0.1%-4.1%) for the standard care group. ● Post hoc sensitivity analyses of the primary end point adjusting for day 1 clinical status score and symptom duration, imputing missing patients as dead, and using the ITT population produced significant results for the 5-day course compared with standard care, but no difference between the 10-day course and standard care.
● Overall mortality, initiation of ventilation, and duration of hospital stay (SOLIDARITY trial end point): Remdesivir did not reduce in-hospital mortality (death rate ratio, 0.95 [95% CI, 0.81-1.11]; P = .5), initiation of mechanical ventilation (295 patients receiving remdesivir vs 284 patients receiving control), or hospitalization duration compared with standard of care. Mortality also did not differ with remdesivir compared with its control when analyzed by ventilator status (ventilated vs not ventilated) or when assessed as a composite end point with initiation of ventilation. ● Clinical status at day 15 on the 7-point ordinal scale (DisCoVeRy add-on study end point): Results are not available.
● Other outcomes for the add-on study for which results are not yet reported include the following: ● Time to improvement of 1 category on ordinal scale from admission. ● Time to discharge or to a National Early Warning Score of 2 or less maintained for 24 hours, whichever occurs first. ● Oxygen supplementation–free days in the first 28 days. ● Ventilator-free days in the first 28 days. ● Incidence and duration of new mechanical ventilation use. ● Duration of hospitalization.
Contraindications, Warnings, and Precautions
Contraindications
Remdesivir is contraindicated in patients with a history of clinically significant hypersensitivity reactions to remdesivir or to any component of the formulation (betadex sulfobutyl ether sodium and possibly hydrochloric acid and/or sodium hydroxide). 1
Warnings and Precautions
Hypersensitivity reactions, including infusion-related and anaphylactic reactions, have been observed during and following remdesivir administration. Signs and symptoms may include hypotension, hypertension, tachycardia, bradycardia, hypoxia, fever, dyspnea, wheezing, angioedema, rash, nausea, diaphoresis, and shivering. Slowing of the infusion rate, with a maximum infusion time of up to 120 minutes, may be considered to prevent these signs and symptoms. Patients should be monitored closely for signs and symptoms of hypersensitivity during and following remdesivir administration. If clinically significant signs and symptoms of hypersensitivity occur, remdesivir administration should be immediately discontinued and appropriate treatment initiated. 1
Transaminase elevations have been observed in healthy volunteers who received remdesivir as well as in patients with COVID-19 treated with remdesivir. Transaminase elevations have also been reported as a clinical feature of COVID-19, making discernment of the contribution of remdesivir to transaminase elevations in patients with COVID-19 difficult. Hepatic laboratory testing should be conducted in all patients prior to initiating therapy with remdesivir and during treatment as clinically appropriate. Discontinuation of remdesivir should be considered if ALT levels increase to greater than 10 times the ULN or if ALT elevation is accompanied by signs or symptoms of liver inflammation. 1
Coadministration of chloroquine phosphate or hydroxychloroquine sulfate with remdesivir is not recommended. Cell culture data demonstrated an antagonistic effect of chloroquine on the intracellular metabolic activation and antiviral activity of remdesivir. 1
Renal function should be evaluated prior to initiating therapy with remdesivir and while receiving remdesivir as clinically appropriate. The pharmacokinetics of remdesivir have not been evaluated in patients with renal impairment; however, patients with an estimated glomerular filtration rate (eGFR) of 30 mL/minute or greater have received remdesivir for the treatment of COVID-19 with no dosage adjustment. The excipient betadex sulfobutyl ether sodium is renally cleared and accumulates in patients with renal impairment; therefore, administration of remdesivir is not recommended in patients with eGFR less than 30 mL/minute. 1
There are no adequate and well-controlled studies of remdesivir in pregnant women; data from case reports and compassionate use are insufficient to evaluate drug-associated risk. In nonclinical reproductive toxicity studies, no adverse effects on embryo-fetal development were observed at systemic exposures (AUC) of the predominant circulating metabolite of remdesivir (GS-441524) that were 4 times (rats and rabbits) the exposure in humans at the recommended human dose. Pregnant women hospitalized with COVID-19 are at risk for serious morbidity and mortality. 1 A study is being conducted to evaluate the pharmacokinetics and safety of remdesivir in pregnant patients with COVID-19. 7
There are no data regarding the presence of remdesivir in human milk, or its effects on breastfeeding infants or milk production. The benefits of breastfeeding should be considered along with the mother’s clinical need for remdesivir and any potential adverse effects on the breastfeeding child from remdesivir or the underlying maternal condition. Breastfeeding individuals with COVID-19 should follow recommendations to avoid exposing the infant to COVID-19. 1
Safety and effectiveness of remdesivir in the treatment of COVID-19 have been established in pediatric patients 12 years and older and weighing at least 40 kg. Safety and effectiveness of remdesivir have not been established in pediatric patients younger than 12 years or weighing less than 40 kg. 1 A study is being conducted to evaluate the safety, tolerability, pharmacokinetics, and treatment response to remdesivir in pediatric patients from birth to younger than 18 years with COVID-19. 7 An EUA allows for emergency use for the treatment of suspected or laboratory-confirmed COVID-19 in hospitalized pediatric patients weighing at least 3.5 kg.2,3
Adverse Reactions
The most common adverse reactions observed with remdesivir (incidence of at least 5%) have included nausea, increased ALT, and increased AST. Less frequent adverse reactions have included hypersensitivity reactions, generalized seizure, and rash. In a placebo-controlled trial (ACTT-1), severe reactions (grade 3 or higher) occurred in 8% of patients treated with remdesivir and in 9% treated with placebo. Serious reactions occurred in 0.4% of patients treated with remdesivir and in 0.6% treated with placebo; serious reactions in the remdesivir group were seizure and infusion-related reaction. Adverse reactions leading to discontinuation of treatment occurred in 2% of remdesivir-treated patients and in 3% of placebo-treated patients; these reactions in the remdesivir group included seizure, infusion-related reaction, increased transaminases, increased ALT and AST, decreased GFR, and acute kidney injury. Adverse reactions reported in open-label trials were similar to those observed in the placebo-controlled trial. 1
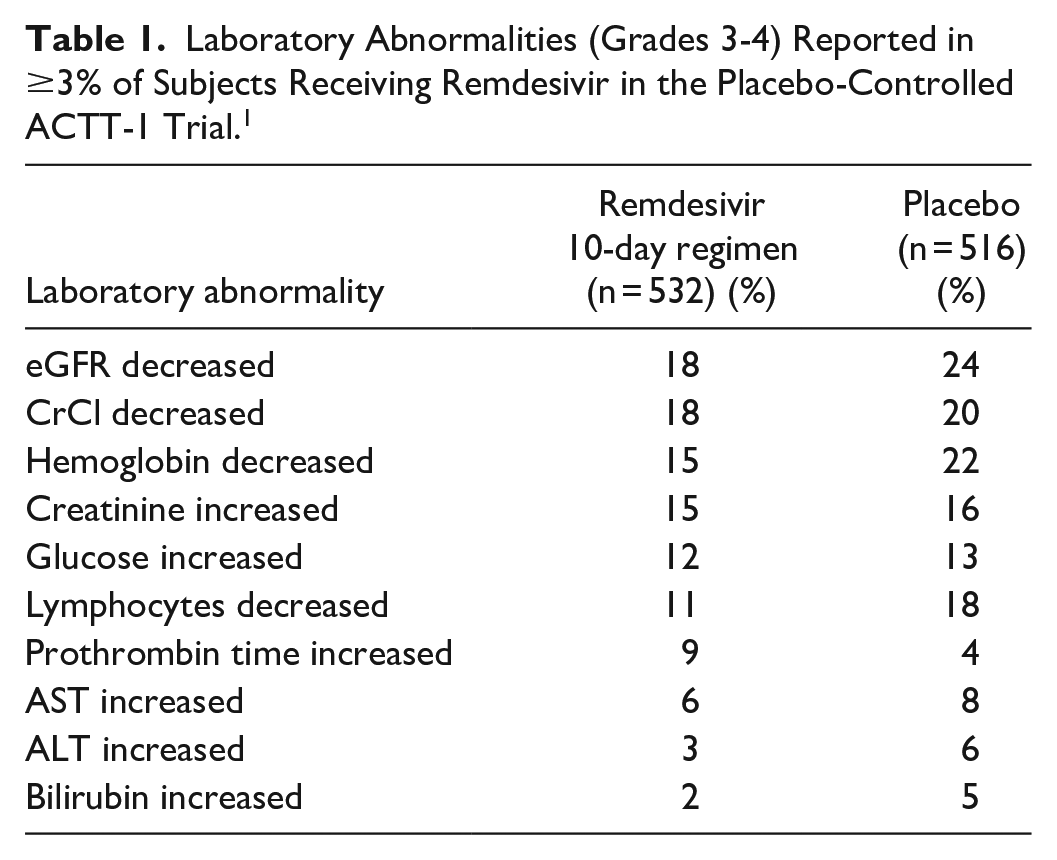
Grade 3 and 4 laboratory abnormalities observed in the phase 3, placebo-controlled ACTT-1 trial are summarized in Table 1. 1
Laboratory Abnormalities (Grades 3-4) Reported in ≥3% of Subjects Receiving Remdesivir in the Placebo-Controlled ACTT-1 Trial. 1
Drug Interactions
Concomitant use of chloroquine phosphate or hydroxychloroquine sulfate with remdesivir is not recommended, due to antagonistic effects observed in cell culture. 1
No formal drug interaction studies have been conducted in humans. In vitro, remdesivir is a substrate of CYP3A4 and OATP1B1 and P-gp transporters and an inhibitor of CYP3A4, OATP1B1, OATP1B3, and MATE1. The GS-704277 metabolite is a substrate for OATP1B1 and OATP1B3. The clinical significance of these in vitro observations is unknown. 1
Recommended Monitoring
Baseline and during remdesivir administration when clinically appropriate: Hepatic function tests (ALT, AST, bilirubin, alkaline phosphatase, prothrombin time); renal function tests (serum creatinine, CrCl); signs/symptoms of infusion reaction. Prothrombin time should also be assessed. 1
Dosing
Prior to initiating therapy and as clinically indicated during therapy, eGFR and hepatic laboratory testing and prothrombin time should be assessed. 1
The recommended remdesivir dosage for adults and pediatric patients 12 years and older and weighing at least 40 kg is a single loading dose of 200 mg on day 1 via IV infusion, followed by once-daily maintenance doses of 100 mg from day 2 via IV infusion. Therapy should be continued for 5 days in patients not requiring invasive mechanical ventilation and/or ECMO but may be extended up to an additional 5 days if a patient does not exhibit clinical improvement, for a total treatment duration of 10 days. The recommended treatment duration in patients requiring invasive mechanical ventilation and/or ECMO is 10 days. 1
Remdesivir is not recommended in patients with eGFR less than 30 mL/minute. 1
Remdesivir must be diluted prior to IV infusion and must be prepared and administered under the supervision of a health care provider. The lyophilized powder must be reconstituted with sterile water for injection and then diluted in a 100 mL or 250 mL sodium chloride 0.9% infusion bag. The injection solution must be diluted in a 250 mL sodium chloride 0.9% infusion bag. Refer to product labeling for reconstitution and dilution instructions. Remdesivir should be administered via IV infusion over 30 to 120 minutes. 1
The EUA provides for emergency use of remdesivir lyophilized powder for treatment of suspected or laboratory-confirmed COVID-19 in hospitalized pediatric patients weighing at least 3.5 kg. The recommended dosage for pediatric patients weighing 3.5 to less than 40 kg is a single loading dose of 5 mg/kg on day 1 followed by 2.5 mg/kg once daily from day 2. The recommended dosage for pediatric patients younger than 12 years and weighing 40 kg or more is a single loading dose of 200 mg on day 1 followed by once-daily maintenance doses of 100 mg from day 2. 2 Infusions should be administered over 30 to 120 minutes, and the recommended duration of therapy is the same as for patients 12 years and older.1,2 Use is not recommended in pediatric patients older than 28 days with eGFR less than 30 mL/minute or in full-term neonates at least 7 to 28 days of age with serum creatinine 1 mg/dL or greater. 2
Product Availability and Storage
Remdesivir was approved by the FDA on October 22, 2020. 7 It is available as a 100 mg single-dose vial containing sterile, preservative-free lyophilized powder and as a 100 mg per 20 mL (5 mg/mL) single-dose vial containing a sterile, preservative-free solution for injection. 1
The vials containing lyophilized powder should be stored below 30°C (86°F) until required for use. Once reconstituted, vials should be used immediately to prepare the diluted solution. The vials containing 100 mg per 20 mL of injection solution should be refrigerated at 2 to 8°C (36 to 46°F). Diluted remdesivir solution in infusion bags may be stored for up to 24 hours at room temperature (20 to 25°C [68 to 77°F]) prior to administration or for 48 hours refrigerated (2 to 8°C [36 to 46°F]). 1
Drug Safety/REMS
No REMS is required for remdesivir. 7
Conclusion
Remdesivir is an antiviral that is FDA approved for the treatment of COVID-19 requiring hospitalization in adults and pediatric patients 12 years and older and weighing at least 40 kg. An EUA also provides for emergency use in hospitalized pediatric patients weighing at least 3.5 kg. Remdesivir has been shown to improve clinical recovery time in patients with severe disease; impact on survival has not been shown. Guideline recommendations regarding remdesivir use vary by organization and are evolving as new data become available. Additional studies are necessary to further identify the patient populations most likely to benefit from therapy, optimal timing for the initiation of therapy, and the role of remdesivir in conjunction with other therapeutics, including dexamethasone and monoclonal antibodies.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.