
Abstract
Diabetes mellitus is one of the most common metabolic diseases worldwide, with periodontal tissue destruction being a major complication. Hyperglycemia-induced changes in metabolism and immune responses may lead to persistent periodontal tissue destruction. This study aimed to investigate hyperglycemia-induced chronic periodontal tissue destruction by focusing on dihydrofolate reductase (DHFR) and its role in metabolic memory. We used CD45.2+ BKS-Leprem2Cd479/Gpt mice and CD45.1+ FVB/NJGpt mice to construct metabolic memory and bone marrow transplantation models, respectively. Our findings showed that hyperglycemia induced a persistent inflammatory senescent phenotype in macrophages. Insulin glycemic control was unable to reverse these pathological changes in bone marrow–derived macrophages and gingival tissues. Furthermore, combined metabolomic and transcriptomic analyses revealed reduced DHFR-mediated 1-carbon metabolism under hyperglycemia, with decreased levels of the reduced form of nicotinamide-adenine dinucleotide phosphate and adenosine triphosphate caused by altered glucose metabolism, impairing the function of DHFR. Alterations in DNA methylation may be responsible for memory-like metabolic patterns in macrophages. Finally, DHFR overexpression reversed hyperglycemia-induced persistent metabolic and pathological changes in macrophages. In summary, this study highlights DHFR-mediated metabolic memory in macrophages as a key factor driving hyperglycemia-induced chronic periodontal tissue destruction.
Keywords
Introduction
Periodontitis is a chronic inflammatory disease that primarily affects the supporting tissues of teeth (Hajishengallis and Chavakis 2021) and is the leading cause of tooth loss (Genco and Sanz 2020). Although periodontitis is primarily caused by bacterial infections, increasing evidence links periodontal diseases such as alveolar bone destruction, gingival fiber destruction, and inflammatory cell infiltration to systemic health. Periodontitis complicates glycemic control (Genco et al. 2020) and contributes to chronic inflammatory responses (Kuraji et al. 2021). Conversely, hyperglycemia alters immune responses, increasing the risk of periodontitis (Polak et al. 2020). More importantly, diabetes mellitus causes specific metabolic and pathological changes, making the control and treatment of periodontitis more challenging.
Diabetes mellitus significantly impairs the immune response (Herold et al. 2024), and prolonged exposure to hyperglycemia has long-term effects on periodontal health (Graves et al. 2020). Despite glycemic control, metabolic changes may persist, increasing the risk of certain comorbidities, including periodontal tissue destruction (Dong et al. 2024). This indicates the overlooked mechanisms underlying diabetes mellitus. Metabolic memory, a phenomenon in which hyperglycemia induces persistent pathological changes, is influenced by early glycemic and glycated hemoglobin levels (Diabetes Control and Complications Trial [DCCT]/Epidemiology of Diabetes Interventions and Complications [EDIC] Study Research Group 2016). Understanding the effects of metabolic memory may provide new insights into managing hyperglycemia-induced chronic periodontal tissue destruction.
Macrophages are promising targets for preventing and treating periodontal tissue destruction (Rattanaprukskul et al. 2024). Macrophages maintain tissue homeostasis by regulating inflammatory responses and facilitating tissue repair (Park et al. 2022). However, macrophages undergo significant changes in diabetes mellitus, influencing various comorbidities such as periodontal tissue destruction (Kuznetsova et al. 2020). Classically activated macrophages (M1 macrophages) rely primarily on aerobic glycolysis (Li et al. 2023). During chronic inflammation, macrophages may sustain inflammatory responses through metabolic memory (Hsu et al. 2023). Excessive glycolysis and activation of inflammatory pathways in diabetes mellitus drive tissue damage (Zhu et al. 2022). Nuclear factor kappa-B (NF-κB) activation contributes to tissue damage through the secretion of downstream inflammatory cytokines (Ko et al. 2019; Locati et al. 2020). In addition, diabetes mellitus induces macrophage senescence, accompanied by the release of proinflammatory factors (Aquino-Martinez 2023).
However, molecular mechanisms underlying metabolic memory remain controversial. Studies have shown that epigenetics significantly regulates deoxyribonucleic acid (DNA) expression and maintains functional changes (Kuraji et al. 2021). Changes in DNA methylation may affect macrophage function by driving metabolic memory. One-carbon metabolism, which requires a reduced form of nicotinamide-adenine dinucleotide phosphate (NADPH) and adenosine triphosphate (ATP), involves the conversion and use of single-carbon units for biosynthesis and energy metabolism (Gan et al. 2024), and these single-carbon units are essential for various methylation reactions (Parsa et al. 2020). Dihydrofolate reductase (DHFR) is crucial for providing single-carbon units required for the synthesis of methylation donors and plays a key role in the 1-carbon metabolism process (Zhao et al. 2021). However, the molecular mechanisms by which hyperglycemia triggers metabolic memory via DHFR-mediated 1-carbon metabolism remain unclear (Nayak et al. 2021). This study, therefore, investigates the role of DHFR in metabolic memory and its effect on sustained pathological changes in periodontal tissue destruction.
Materials and Methods
Study Design
In this study, 2 animal models were used: (1) a metabolic memory model constructed by CD45.2+ BKS-Leprem2Cd479/Gpt mice (N group: mice with normal glycemia; D group: mice with 12 wk of diabetes; Ins group: mice received 8 wk of insulin glycemic control after 12 wk of diabetes; 2-DG group: mice received 8 wk of 2-deoxy-D-glucose [2-DG] treatment after 12 wk of diabetes) and (2) a bone marrow transplantation model constructed by CD45.1+ FVB/NJGpt mice (FVB-T group: mice received bone marrow transplantation from CD45.2+ BKS-Leprem2Cd479/Gpt mice; FVB-C group: control group, mice received bone marrow transplantation from CD45.1+ FVB/NJGpt mice). The animal experiment conformed with the ARRIVE guidelines. Please refer to the appendix file for detailed animal experiment design and modeling methods (Appendix Fig. 1).
A corresponding in vitro model was constructed by immortalized bone marrow–derived macrophages (iBMDMs). The LG group served as a control and was cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 25 mM glucose for 72 h, the HG group was cultured in DMEM containing 45 mM glucose for 72 h, the H-L group was cultured with DMEM containing 45 mM glucose for 72 h and then with DMEM containing 25 mM glucose for another 72 h, and the 2-DG group was cultured with DMEM containing 45 mM glucose for 72 h and then with 2 mM 2-DG for another 24 h. Please refer to the appendix file for the detailed cell experiment design and modeling methods.
DHFR Overexpression
The DHFR overexpression plasmid constructed with pcDNA3.1 backbone was purchased from Hanbio. The reconstructed plasmid and control vectors were transfected into iBMDM cells using LipoFiter (HB-LF-1000, Hanbio) following the manufacturer’s instructions. After incubation for 24 h, the medium was removed, and the infected cells were incubated under the culture conditions described above. Please refer to the appendix file for detailed DHFR overexpression experiment grouping, methods, and specific methods of other experiments used in this study.
Statistical Analysis
Data were analyzed using GraphPad Prism software (version 10.1.1, GraphPad Software, LLC) and presented as mean ± standard deviation. For experiments with more than 3 groups, 1-way analysis of variance and Tukey’s multiple comparison method were used to compare the differences between groups. For experiments involving 2 groups, an unpaired t test was performed to determine the differences. Asterisks represent all significant comparisons. Statistical significance was set at P < 0.05.
Results
High-Glucose Environment Induces Metabolic Memory
We first investigated the metabolic memory effect induced by hyperglycemia. Insulin treatment restored fasting blood glucose (FBG), food intake, water consumption, and urine and feces output to levels comparable with those of control mice (Appendix Fig. 2A). Western blot analysis of gingival tissue and BMDMs of 12-wk-old diabetic mice showed elevated glycolytic enzyme levels and decreased tricarboxylic acid cycle enzymes, indicating a shift toward aerobic glycolysis. Notably, insulin glycemic control failed to reverse these alterations, suggesting that metabolic memory was established after 12 wk of hyperglycemia (Appendix Fig. 2B, C). Thus, 12-wk-old mice were selected for subsequent experiments.
Correlation analysis of all differentially expressed metabolites (DEMs) in gingival tissue revealed the highest correlation between the D and Ins groups, suggesting that insulin treatment did not fully reverse hyperglycemia-induced metabolic changes (Fig. 1A). Heatmap analysis of DEMs in BMSCs and BMDMs showed that diabetic BMDMs had a significantly different metabolic profile (Fig. 1B). Principal component analysis of all DEMs of gingiva and cells revealed that the BMDM, Ins, and D groups were similar, highlighting the metabolic memory induced by hyperglycemia (Fig. 1C). Further, immunohistochemistry analyses revealed that hyperglycemia-induced expression of gingival hexokinase 2 (HK2) and pyruvate kinase M2 (PKM2) in gingival tissue could not be reversed by insulin treatment alone (Fig. 1D, E), suggesting a role for metabolic memory.
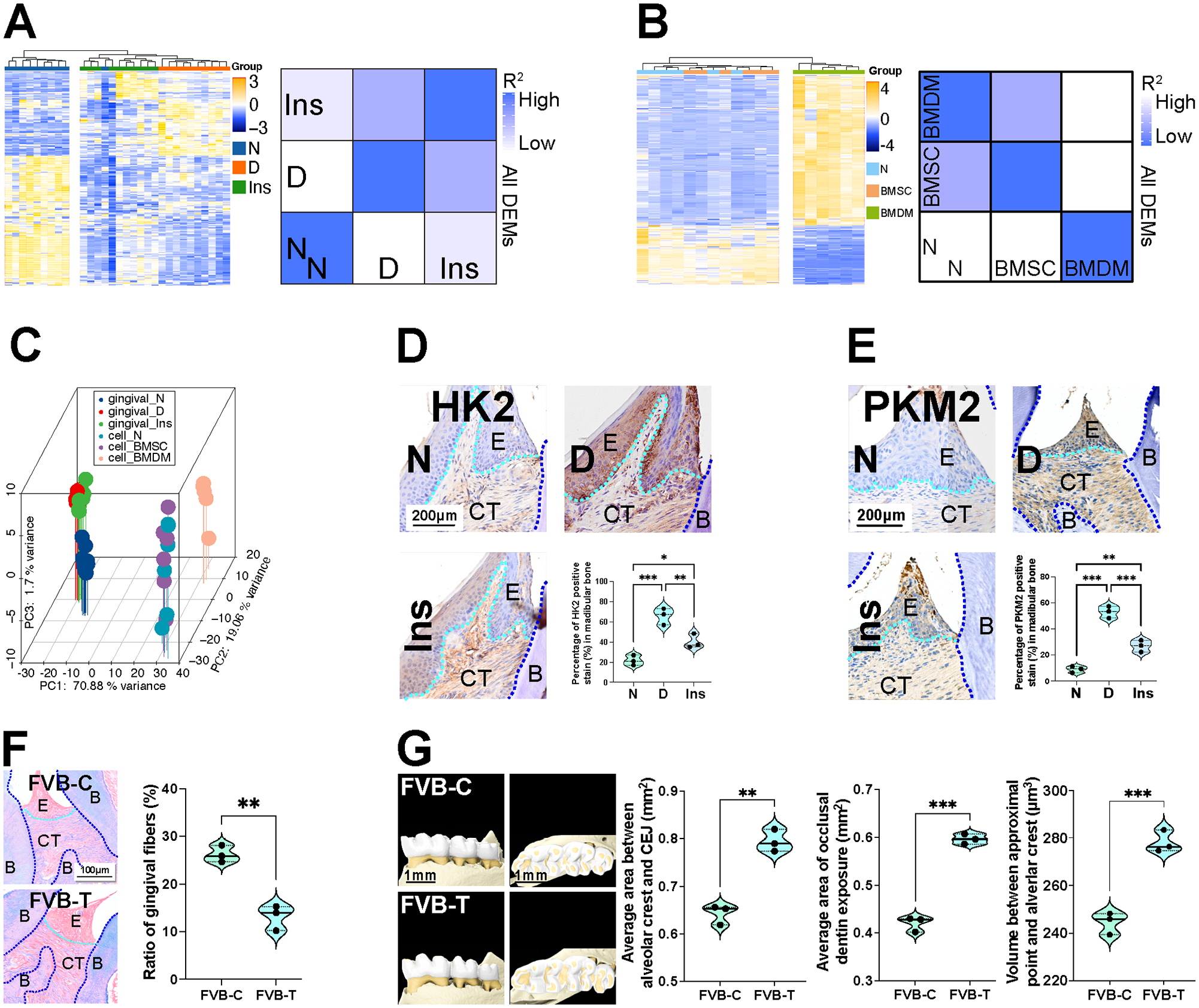
Hyperglycemia causes metabolic memory. Bone marrow–derived macrophages (BMDMs) and gingiva were collected from metabolic memory mice model for quasi-targeted metabolomic analysis. After, 6-wk-old FVB/NJGpt mice were given whole-body X-ray irradiation before bone marrow transplantation to search pathological changes in periodontitis. (
Bone marrow–transplanted mice were constructed to further investigate the development of metabolic memory. Normal FBG levels were observed in the FVB-T and FVB-C groups (Appendix Fig. 3A). Flow cytometry confirmed the presence of donor CD45.2+ immune cells in recipient mice (Appendix Fig. 3B). Despite normal FBG levels, FVB-T mice exhibited increased expression of glycolytic proteins in gingival tissue (Appendix Fig. 3C). Immunofluorescence showed that macrophages with high HK2 expression were present in the gingival tissues of recipient mice (Appendix Fig. 3D). Inflammatory cell infiltration, osteoclast activation (Appendix Fig. 3E), and fiber destruction (Fig. 1F) were also observed in FVB-T gingiva. Micro–computed tomography (micro-CT) analysis showed slight but statistically significant alveolar bone destruction in FVB-T mice. Specifically, in FVB-C mice, the average area between the alveolar crest and the cementoenamel junction (CEJ), the area of dentin exposed on the occlusal surface of the mouse molars (occlusal dentin exposure area), and the volume between the contact point and the alveolar crest were 0.64 mm2, 0.42 mm2, and 244.5 µm3 respectively. In FVB-T mice, the values were 0.79 mm2, 0.60 mm2, and 278.2 µm3, respectively (Fig. 1G).
Glycolysis Inhibition Alleviates Persistent Macrophage Dysfunction
To investigate the molecular mechanisms, we conducted cell experiments to study the impact of high glucose on macrophage function. Gingival single-cell RNA sequencing (scRNA-seq) was performed to identify the major cells involved in hyperglycemia-induced periodontitis. Although the number and proportion of macrophages were reduced in diabetes (Appendix Fig. 4A), cell–cell communication analysis revealed increased overall interaction frequency among gingival cell types (Appendix Fig. 4B). Moreover, the network plot of interaction strength showed the highest connections between macrophages and other cell types, hinting that macrophages exhibited the strongest interaction strength (Fig. 2A). Differentially expressed genes (DEGs) of different cells in the gingiva demonstrated that macrophages were involved in inflammatory and cellular senescence pathways (Fig. 2B). Macrophage migration inhibitory factor and cathepsin G, key factors regulating inflammation, were upregulated in diabetes. Meanwhile, the C-X-C motif ligand, colony-stimulating factor, and platelet-derived growth factor, which were involved in the macrophage function, were downregulated in diabetes (Appendix Fig. 4C). These findings suggest that macrophages may play a critical role in the development of hyperglycemia-induced chronic periodontal tissue destruction.
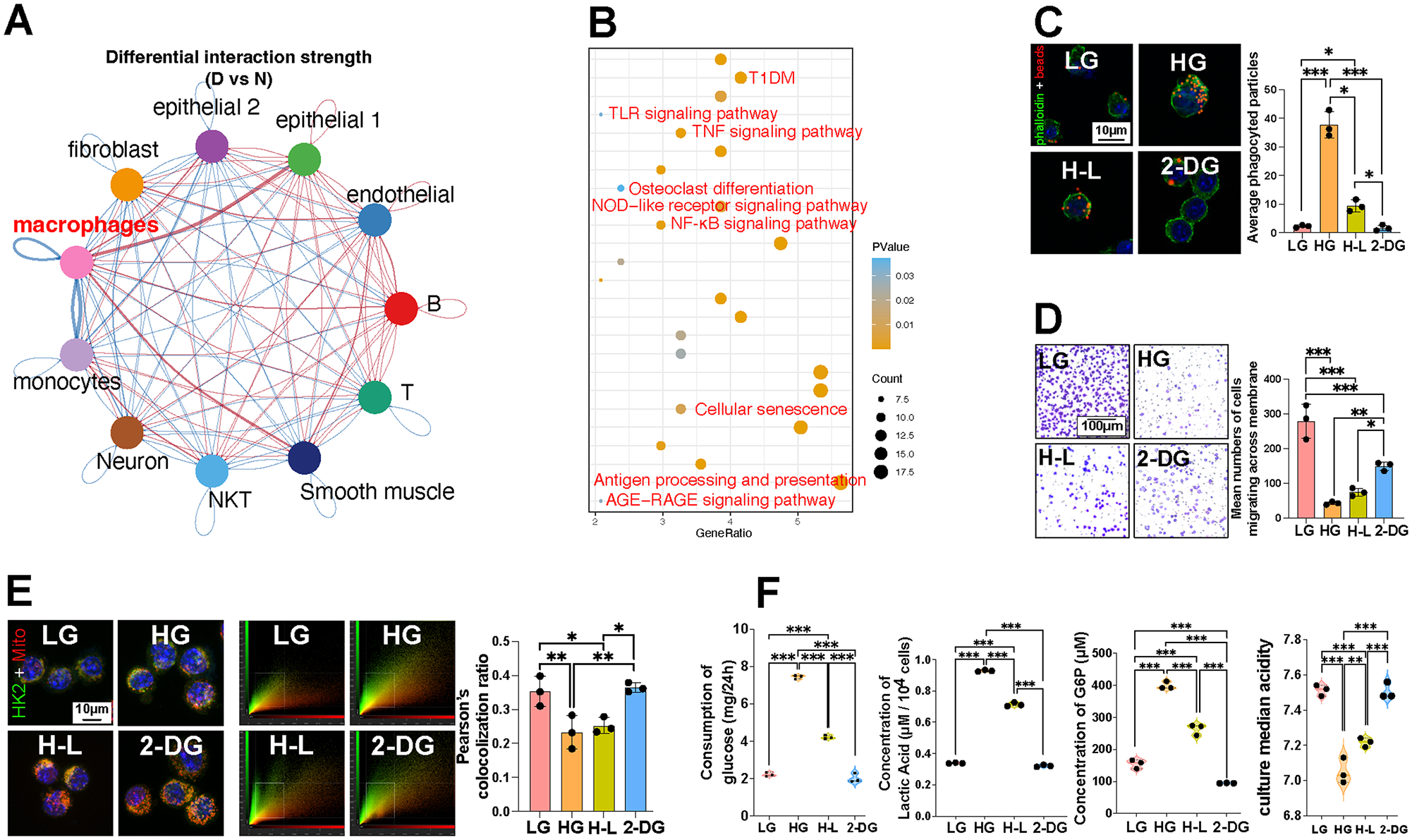
Inhibition of glycolysis alleviates persistent macrophage dysfunction induced by high-glucose.
To explore the influence of glycolysis on metabolic memory, we investigated macrophage function after treatment with 2-DG. Our data showed persistent alterations in phagocytosis (Fig. 2C, Appendix Fig. 5A), chemotaxis (Fig. 2D), senescent phenotype, and antigen presentation capability despite switching back to a normal glucose medium (Appendix Fig. 5B, C). Since major histocompatibility complex II (MHC II) is a marker of M1 macrophages (Dechantsreiter et al. 2022), increased MHC II and decreased CD163 levels indicated a permanent shift toward the M1 phenotype under high glucose levels (Appendix Fig. 5D, E). These changes were further reduced by inhibiting glycolysis with 2-DG.
Given the close link between macrophage function and aerobic glycolysis, we examined the expression of key enzymes. Transferring cells back to normal glucose did not fully reverse altered expression levels (Appendix Fig. 5F). We also assessed HK2 and DNA methyltransferase 1 (DNMT1) expression in macrophages and found that returning them to normal glucose did not restore this alteration (Appendix Fig. 5G, H). Colocalization analysis of HK2 and mitochondria revealed a persistent decrease in the colocalization coefficient, indicating prolonged proinflammatory activity (Fig. 2E). The analysis of glycolytic metabolism in macrophages showed that glycolysis was permanently influenced by high-glucose conditions, with a lower culture median acidity (7.05) and a higher glucose consumption (7.433 mg/24 h) and glucose-6-phosphatase concentration (398.5 µM) in the HG group (Fig. 2F). These changes were reversed by inhibiting glycolysis with 2-DG.
Glycolysis Inhibition Alleviates Persistent Periodontal Inflammatory Damages
To validate the systemic effects of metabolic memory through glycolysis inhibition experiments, we used our animal models (Appendix Fig. 6A). To rule out potential confounding effects of leptin receptor deficiency on our results, we examined HK2 and DNMT1 expression in the gingival tissue of BKS-Leprem2Cd479/Gpt mice before glycemia elevated. Our findings showed no statistically significant difference in HK2 and DNMT1 expression as compared with the control group (Appendix Fig. 6B). This suggests that hyperglycemia-induced glycolysis abnormalities and DNA methylation changes were independent of leptin receptor deficiency. Furthermore, since the occurrence of metabolic memory primarily depends on the effect of a hyperglycemic environment on cellular metabolism and epigenetics, this result also indicates that hyperglycemia-induced metabolic memory and persistent pathological changes in periodontitis are not influenced by leptin receptor deficiency. Gingival protein analysis revealed that persistent metabolic alterations caused by hyperglycemia were resolved only when glycolysis was inhibited with 2-DG (Appendix Fig. 6C).
Micro-CT revealed that alveolar bone resorption persistently increased in diabetic mice despite glycemic control. The average area between the alveolar crest and CEJ, occlusal dentin exposure area, and volume between the contact point and the alveolar crest in group N were 0.32 mm2, 0.14 mm2, and 253.7 µm3 respectively. For group D, the above-mentioned data were 1.40 mm2, 0.70 mm2, and 722.0 µm3 respectively (Fig. 3A). Prolonged infiltration of inflammatory immune cells (Fig. 3B), increased active osteoclasts (Fig. 3C), fiber destruction (Fig. 3D), and increased DNMT1 expression (Fig. 3E) were also observed in the gingiva. Even after achieving glycemic control, these inflammatory pathological changes persisted until glycolysis was inhibited by 2-DG treatment.
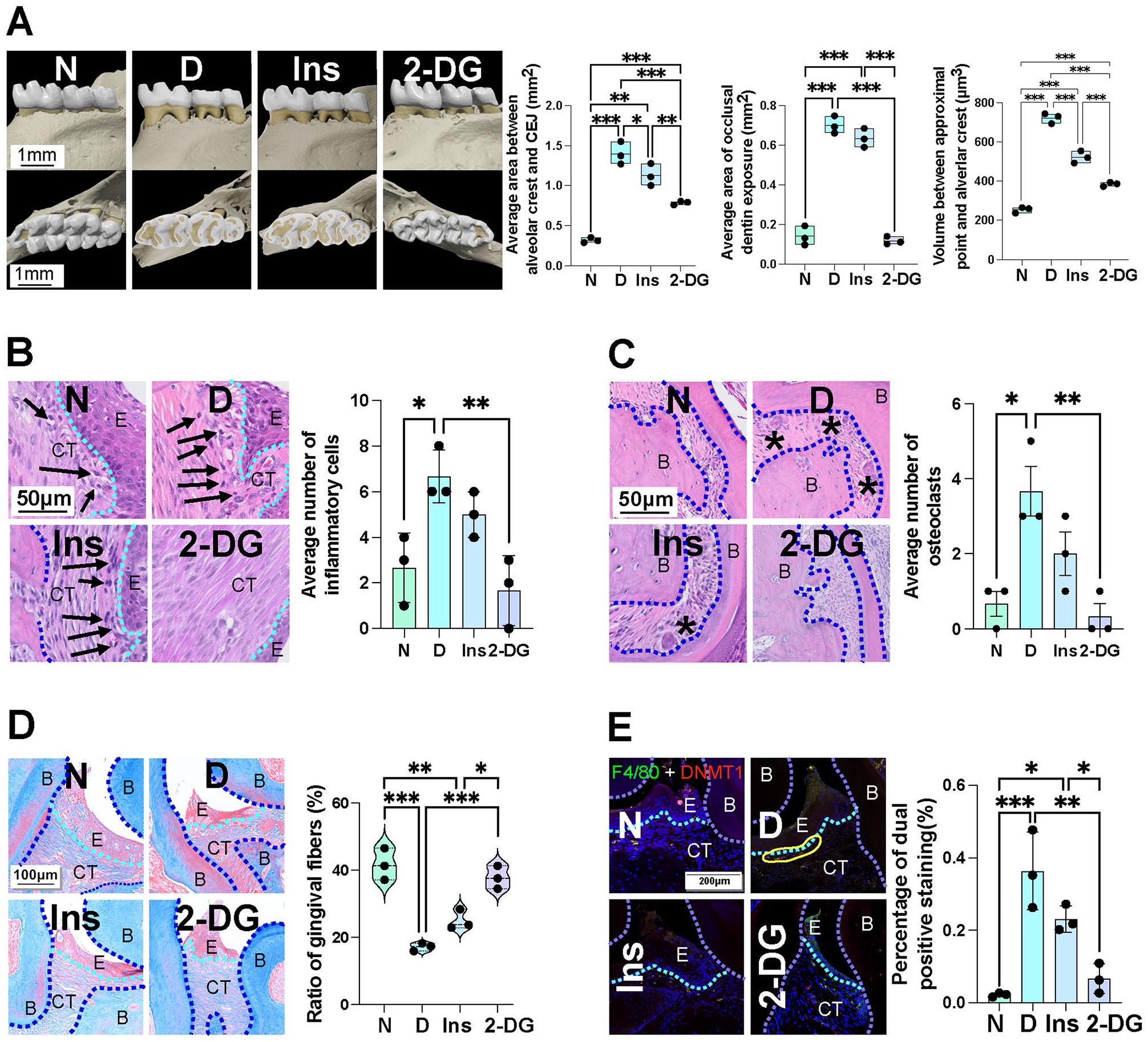
Inhibition of glycolysis alleviates persistent periodontal tissue destruction induced by diabetes. A metabolic memory mice model was used to further confirm the influence of glycolysis on metabolic memory. (
DHFR-Mediated 1-Carbon Metabolism Sustains Metabolic Memory
To understand why alterations in glucose metabolism cause metabolic memory, we further investigated their relationship (Appendix Fig. 7A). Kyoto Encyclopedia of Genes and Genomes analysis of DEMs in the gingiva and BMDMs revealed major alterations in the metabolic pathways related to 1-carbon metabolism (Fig. 4A, Appendix Fig. 7B). Based on these findings, we characterized the metabolites involved in 1-carbon metabolism. We observed decreases in serine, glycine, methionine, tetrahydrofolate, S-adenosylhomocysteine (SAH), and S-adenosylmethionine (SAM) (Appendix Fig. 7C). However, the methylation index (SAM/SAH) significantly decreased in bone marrow mesenchymal stem cells (BMSCs) but showed no significant change in BMDMs (Fig. 4B), suggesting that hyperglycemia-induced DNA methylation changes likely occur in BMSCs. Although the methylation index normalized after differentiation into BMDMs, the metabolic and phenotypic alterations triggered by epigenetic changes in the BMSCs stage persisted into BMDMs.
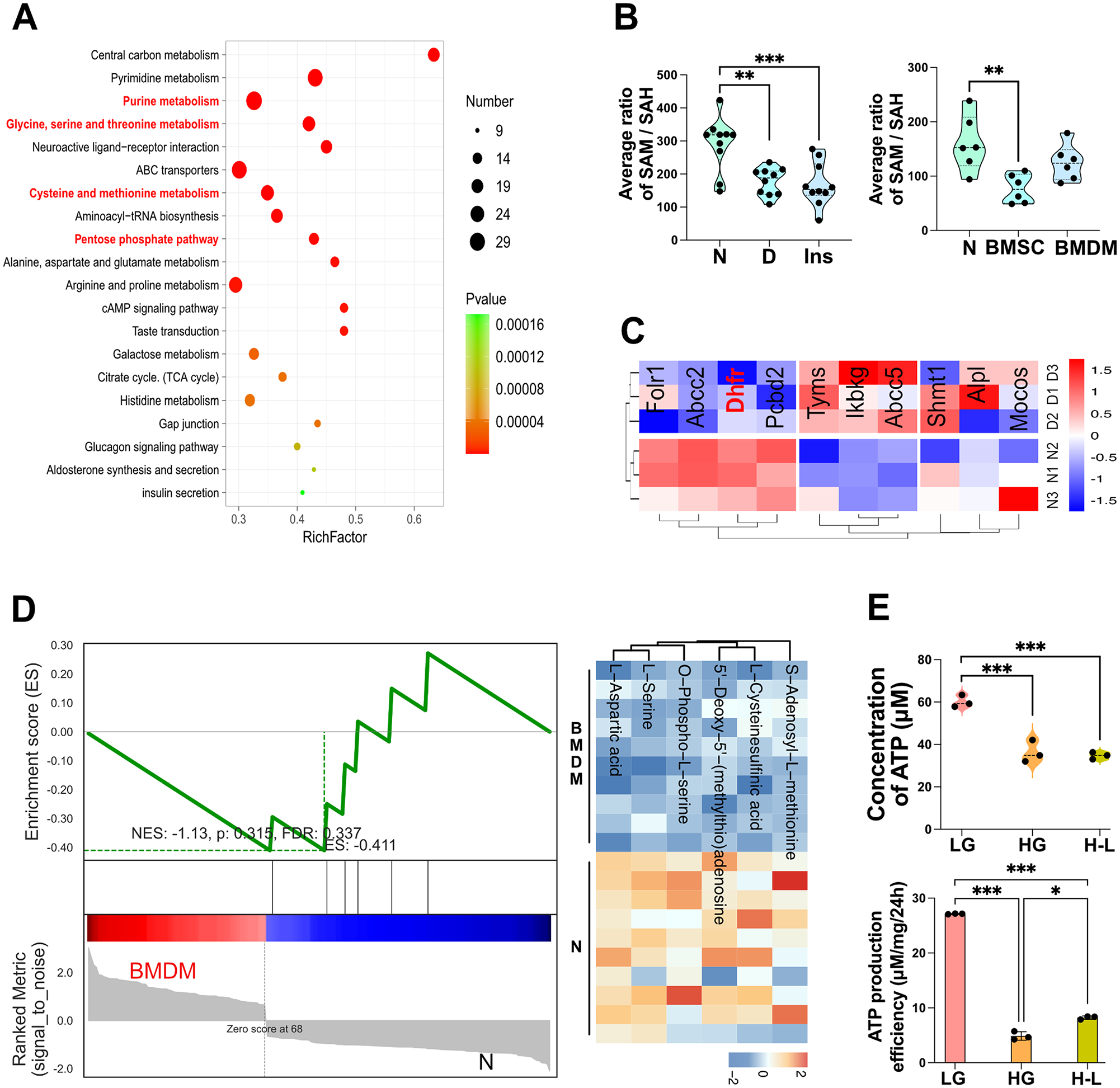
Decreased dihydrofolate reductase (DHFR)–mediated 1-carbon metabolism leads to metabolic memory. Multiomics analysis was performed to find the underlying mechanism of metabolic memory. (
Transcriptomic analysis of BMDMs showed that DEGs were enriched in pathways related to 1-carbon metabolism, including 1-carbon pool by folate and antifolate resistance (Appendix Fig. 7D). DHFR, enriched in both pathways, exhibited decreased expression in diabetes (Fig. 4C). Further analysis of proteins related to glycine, SAM, and methionine metabolism in BMDMs and identified 66, 111, and 124 DEGs, respectively. Among these, 20 proteins, including DHFR, were associated with all 3 metabolites (Appendix Fig. 7E). These findings suggest that DHFR may play a key role in metabolic memory.
Correlation analysis revealed a negative correlation between glycolysis and 1-carbon metabolism in BMDMs (Appendix Fig. 7F), with gene set enrichment analysis demonstrating reduced 1-carbon metabolism (Fig. 4D). We also found that the pentose phosphate pathway was significantly downregulated in macrophages (Appendix Fig. 7G). Glycolysis and the pentose phosphate pathway are important for 1-carbon metabolism as they provide NADPH and ATP. A significantly decreased ATP production efficiency (4.892 µM/mg/24 h) was observed in the HG group (Fig. 4E). These changes suggest that hyperglycemia may impair DHFR function by downregulating NADPH and ATP.
DHFR Overexpression Rescues Macrophage Metabolic Disorders
Hyperglycemia significantly reduced DHFR expression, which could not be reversed by insulin treatment alone (Appendix Fig. 8A). Immunohistochemistry analysis in gingival tissues confirmed that DHFR expression remained low despite insulin treatment (Fig. 5A). Since the catalytic function of DHFR requires NADPH, we measured NADPH levels in macrophages and found a persistent decrease under high-glucose conditions, accompanied by a notable increase in the nicotinamide adenine dinucleotide phosphate (NADP+) to NADPH ratio (Fig. 5B). These findings indicate a persistent impairment of DHFR function under high-glucose conditions.
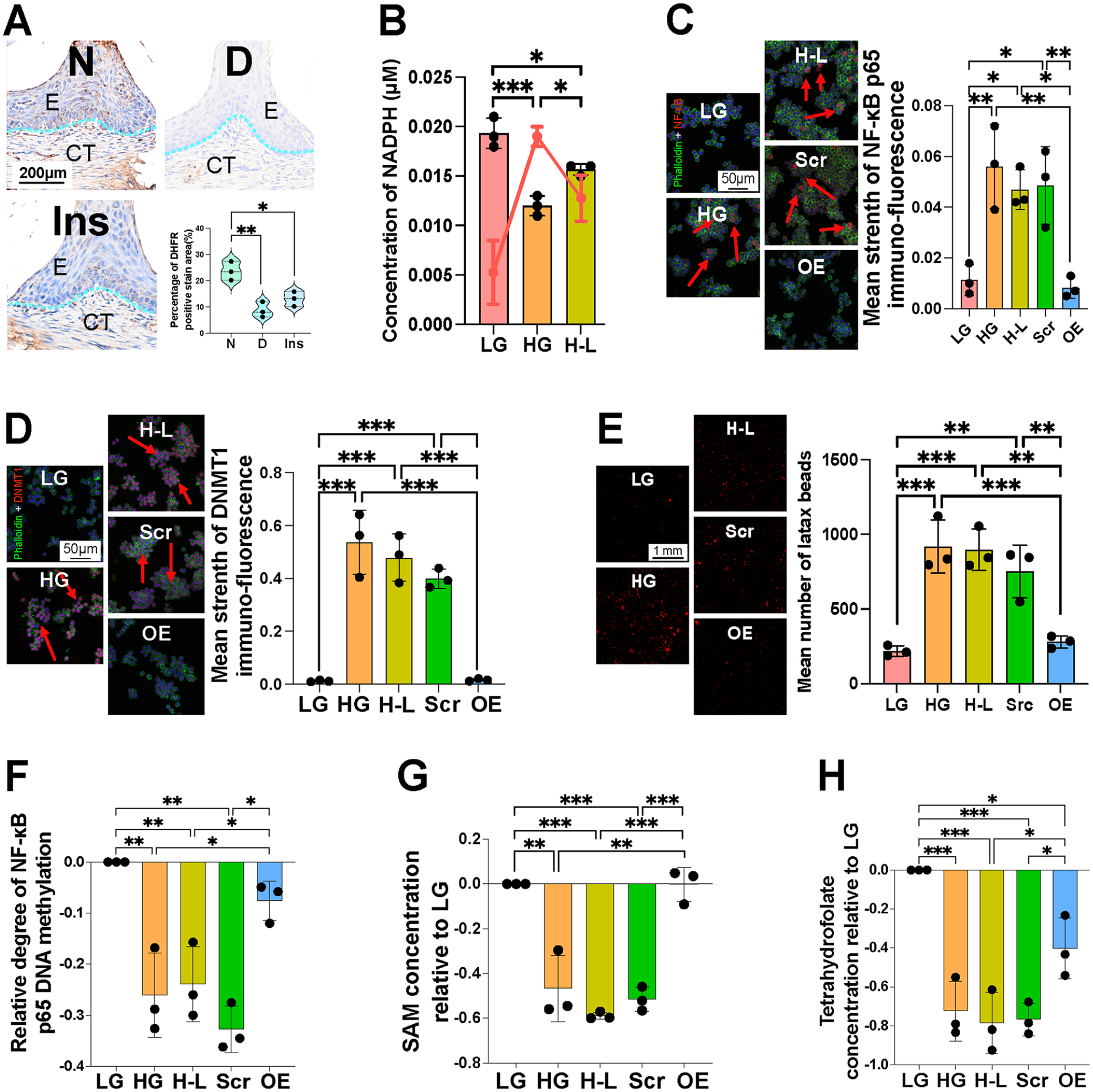
Dihydrofolate reductase (DHFR) overexpression relieved metabolic memory induced by high glucose. Macrophages were transfected with DHFR overexpression plasmid to verify DHFR function. (
Based on these observations, we transfected macrophages with a DHFR overexpression plasmid to elucidate the role of DHFR. Transfection efficiency was verified using green fluorescent protein. DHFR overexpression reversed prolonged DNMT1 and NF-κB p65 activation (Appendix Fig. 8B). This was further corroborated by immunofluorescence (Fig. 5C, D). Further phagocytosis (Fig. 5E), chemotaxis, senescence, and antigen presentation (Appendix Fig. 8C, D, E) tests showed that DHFR overexpression alleviated persistent inflammatory alterations in macrophages caused by high glucose levels. In addition, DHFR overexpression restored the permanently reduced DNA methylation level of NF-κB p65 (Fig. 5F). Notably, these changes were synchronized with alterations in the concentrations of SAM and tetrahydrofolate (Fig. 5G, H).
Discussion
Studies have reported a higher incidence and severity of periodontitis in patients with diabetes mellitus (Polak et al. 2020). This is largely due to the immune cell dysfunction and metabolic disturbances caused by hyperglycemia. In a high-glucose environment, M1 macrophages release proinflammatory cytokines, exacerbating periodontal tissue destruction. In addition, the accumulation of advanced glycation end-products intensifies periodontal inflammation periodontium (Thomas et al. 2024). However, conventional theories of diabetes mellitus face a persistent challenge: despite achieving glycemic control, comorbidities such as periodontal tissue destruction persist, suggesting unrecognized pathological mechanisms (Roden and Shulman 2019; Schwartz et al. 2022). Metabolic memory has bridged some of these theoretical gaps. However, its underlying mechanisms remain controversial.
In this study, we proposed that hyperglycemia induces metabolic memory through DHFR-mediated 1-carbon metabolism, resulting in persistent periodontal tissue destruction (Appendix Fig. 9). Our findings demonstrate that insulin glycemic control cannot reverse persistent periodontal tissue destruction, and we provide evidence linking glucose metabolism to metabolic memory. We identified DHFR as a potential therapeutic target for addressing metabolic memory. NADPH and ATP are essential for DHFR-mediated 1-carbon metabolism (Yu et al. 2019; Zhao et al. 2021). Our results revealed that hyperglycemia-induced glycolysis and pentose phosphate pathway alterations led to decreased NADPH and ATP levels, reducing DHFR expression and function. In addition, our results demonstrated that metabolic alterations induce an acidic environment, which significantly impairs DHFR function by hindering its binding to NADPH (Liu et al. 2014). Conversely, DHFR overexpression alleviated persistent inflammatory damage caused by M1 macrophages under hyperglycemia. These findings suggest that hyperglycemia disrupts DFHR-mediated 1-carbon metabolism, alters DNA methylation status of NF-κB, and triggers persistent pathological changes in periodontium.
Our findings further suggest that hyperglycemia promotes M1 polarization, which enhances immune cell recruitment and pathogen elimination but also triggers proinflammatory cytokine secretion and tissue damage (Locati et al. 2020; Chen et al. 2023). We found that HK2 dissociated from mitochondria under high-glucose conditions. HK2 shifts between the cytoplasmic and mitochondrial-bound states in response to environmental and metabolic pressures (Baik et al. 2023). NF-κB–mediated inflammation is associated with the mitochondrial localization of HK2 (Rabbani and Thornalley 2019). HK2 dissociation from mitochondria promotes oxidative stress and inflammatory responses, which are closely associated with hyperglycemia-induced chronic periodontal tissue destruction (Rabbani and Thornalley 2019; Guo et al. 2022). Metabolic memory prolonged this proinflammatory activity and caused persistent pathological changes in periodontium.
There is a complex interaction between genes and metabolites involved in regulating metabolic memory. Genes encoding key glycolytic enzymes can undergo DNA methylation, affecting cellular metabolism (Dhawan et al. 2015). For example, hexokinase DNA methylation level has been proposed as a biological marker for diabetes mellitus (Wakeling et al. 2022). In addition, studies have shown that hyperglycemia significantly affects oral microbiota composition, increasing susceptibility to periodontal tissue destruction (Sangha et al. 2024). In this study, we observed a decrease in methylation index, indicating global DNA demethylation during hyperglycemia. However, DNMT1 expression was increased, which may be attributed to the positive regulatory influence of lactate (Huang et al. 2020). Furthermore, downstream pathways of 1-carbon metabolism may also contribute to this inconsistency, as reduced SAH under hyperglycemia alleviates its inhibitory effect on DNMT1 (You et al. 2023).
We noticed that some studies proposed that the effects of metabolic memory can be primarily attributed to lower cumulative exposure to HbA1c (Miller and Orchard 2020). In addition, the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study attributed most of the beneficial effects of intensified insulin therapy to reductions in HbA1c (Jacobson et al. 2021). Clinical studies on metabolic memory also commonly stratify patients using HbA1c levels (Ceriello et al. 2023). This theory effectively explains incidence reduction of diabetic comorbidities associated with intensified therapy. However, our study demonstrated that the mechanism underlying metabolic memory involves abnormal DNA methylation, sustained activation of inflammatory signaling, and persistent alterations in immune cell function. These findings suggest that metabolic memory is not merely an effect of cumulative HbA1c exposure but rather a deeper mechanism involving gene expression and metabolic reprogramming.
In conclusion, our study highlights the role of DHFR-mediated 1-carbon metabolism in persistent hyperglycemia-induced chronic periodontal tissue destruction. This finding enhances our understanding of the long-term impact of diabetic complications and offers new directions for future therapeutic strategies. Further clinical trials are necessary to test the hypothesis and to explore the impact of early intervention and effective metabolic control on metabolic memory-mediated complications, including periodontal inflammation.
Author Contributions
L. Nie, contributed to conception, design, data acquisition, analysis and interpretation, drafted and critically revised the manuscript; Y. Sun, H. Dong, M. You, A. Cui, Z. Yue, Q. Lv, N. Ji, contributed to data acquisition, critically revised the manuscript; P. Zhao, contributed to data analysis, critically revised the manuscript; H. Wang, X. Xu, W.K. Leung, contributed to design, critically revised the manuscript; J. Wang, contributed to conception and design, critically revised the manuscript; Q. Wang, contributed to conception, design, data interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345251340632 – Supplemental material for DHFR-Driven Metabolic Memory Sustains Periodontal Tissue Destruction
Supplemental material, sj-docx-1-jdr-10.1177_00220345251340632 for DHFR-Driven Metabolic Memory Sustains Periodontal Tissue Destruction by L. Nie, Y. Sun, H. Dong, M. You, A. Cui, Z. Yue, P. Zhao, Q. Lv, N. Ji, H. Wang, X. Xu, W.K. Leung, J. Wang and Q. Wang in Journal of Dental Research
Footnotes
Acknowledgements
We sincerely thank Prof. Ling Ye from State Key Laboratory of Oral Diseases, Sichuan University, and Prof. Hongbo Hu from Department of Rheumatology and Immunology, West China Hospital, for giving us valuable and critical advice on our whole study. We thank Novogene Co., Ltd. (Beijing, China) for carrying out Quasi-targeted metabolomics. We would like to thank Editage (![]() ) for English language editing.
) for English language editing.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (82470985 to Qi Wang, 82271034 to Jian Wang), Natural Science Foundation of Sichuan Province (2024NSFSC0548 to Qi Wang), Innovative Research Group Project of the Natural Science Foundation of Sichuan Province (2023NSFSC2000 to Jian Wang).
Data Availability Statement
The data supporting the findings of this study are available within the article and its supplementary materials.
A supplemental appendix to this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.