
Abstract
Heterozygous mutations of the gene encoding caspase-8 protease occur in 10% of human head and neck squamous cell carcinomas (HNSCCs). Cell line studies indicate that these mutations block apoptosis induced by death ligands. However, the in vivo role of caspase-8 mutations in the development of HNSCC and their impact on response to immune checkpoint blockade have not been determined. We generated mice with heterozygous, epithelium-specific knock-in of a representative, HNSCC-associated caspase-8 mutation (D305G). The impact of the caspase-8 mutation was assessed following treatment with the carcinogen 4-nitroquinoline-1-oxide (4NQO) in drinking water. Treatment of the D305G caspase-8 mutant mice with 4NQO resulted in a greater number of tongue tumors per mouse and a higher percentage of advanced-stage invasive carcinomas than was observed in 4NQO-treated mice with wild-type caspase-8, and tumors from the mutant mice were more resistant to anti–PD-1. We also engineered the murine oral cancer cell line MOC1 for heterozygous expression of caspase-8 mutations. Tumors generated from these engineered cells in syngeneic, immunocompetent mice demonstrated reduced responsiveness to anti–PD-1, relative to tumors with wild-type caspase-8. Further, the caspase-8 mutant tumors displayed reduced intratumoral and splenic CD8+ T cells and impaired recruitment of monocytes and dendritic cells during PD-1 blockade. Collectively, these findings demonstrate that caspase-8 mutation promotes carcinogen-induced HNSCC development and resistance to anti–PD-1. Investigation of caspase-8 mutations as potential biomarkers of poor response to immunotherapy in patients with HNSCC is warranted.
Keywords
Introduction
Head and neck squamous cell carcinomas (HNSCCs) are responsible for almost 5% of all deaths from cancer worldwide (Barsouk et al 2023; Bray et al 2024; Siegel et al 2024). Treatment of these cancers has historically relied on surgical resection, which can cause disfigurement and difficulties in speech and swallowing, and/or treatment with radiation and conventional chemotherapy drugs, which frequently leads to adverse toxicities (Chow 2020; Johnson et al 2020). The advent of immune checkpoint inhibitors (ICIs) has yielded improvement in clinical outcomes and a reduction in unwanted side effects. In particular, treatment with nivolumab or pembrolizumab, monoclonal antibodies targeting the immune checkpoint protein programmed cell death protein 1 (PD-1), can lead to potent and durable responses against HNSCC (Chow et al 2016; Ferris et al 2016; Seiwert et al 2016; Mehra et al 2018; Burtness et al 2019; Johnson et al 2020). However, only a minority of patients with HNSCC (~15%–20%) demonstrate clinical benefit from nivolumab or pembrolizumab. Efforts to rationally select patients who are likely to benefit from ICI therapies have been severely hampered by a lack of knowledge regarding potential biomarkers of response or resistance.
Characterization of the genomic landscape of HNSCC tumors has provided insight into possible mechanisms of tumor development and drug resistance (Cerami et al 2012; Gao et al 2013; Cancer Genome Atlas 2015). Notably, the gene encoding the intracellular cysteine protease caspase-8 is among the more frequently mutated genes in HNSCC tumors, with heterozygous mutations occurring in approximately 10% of human tumors (Agrawal et al 2011; Stransky et al 2011; Cancer Genome Atlas 2015). Mutations in the caspase-8 gene are scattered throughout the coding region, which encodes a proenzyme protease consisting of a small and large subunit.
Caspase-8 protease mediates apoptosis induced by ligand-activated cell surface death receptors, including the receptors for tumor necrosis factor (TNF), TNF-related apoptosis-inducing ligand (TRAIL), and Fas ligand (Ashkenazi and Dixit 1998; Leonard and Johnson 2018). The activation of caspase-8 following death receptor activation propagates the extrinsic apoptosis pathway, leading to cell death. We previously reported that exogenous overexpression of HNSCC-associated caspase-8 mutations in cell line models blocks apoptosis induction by TRAIL or Fas ligand, indicating that the mutant proteins have the capacity to act as dominant-negative proteins (Li et al 2014; Cui et al 2021). Since effector T cells induce apoptosis in target cells, in part, via death ligand activation of caspase-8 and the extrinsic apoptosis pathway, mutation of caspase-8 may provide tumors a means of immune evasion and resistance to ICIs.
In this report, we determined the impact of a heterozygous caspase-8 mutation on the development of HNSCC tumors in the context of an immunocompetent murine model. We further examined whether heterozygous expression of caspase-8 mutants would alter the sensitivity of HNSCC tumors to ICI treatment. We engineered wild-type, immunocompetent mice for epithelium-specific, heterozygous knock-in of an HNSCC-associated caspase-8 mutation. The mutant mice exhibited increased tumorigenicity relative to mice with wild-type (WT) caspase-8 following treatment with 4-nitroquinoline-1-oxide (4NQO), demonstrating a clear role for caspase-8 mutation in promoting carcinogen-induced HNSCC. We also found that HNSCC tumors expressing heterozygous caspase-8 mutations were resistant to the antitumor effects of anti–PD-1. These findings provide a first-time demonstration that HNSCC-associated caspase-8 mutations promote tumor development in vivo and confer resistance to ICI therapy.
Materials and Methods
Extensive experimental details are presented in the Appendix.
Results
Generation of Mice with Conditional, Tissue-Specific Expression of the D305G Caspase-8 Mutation
Caspase-8 gene mutations in human HNSCC occur as heterozygous mutations. To investigate the role of caspase-8 mutation in the development of HNSCC, we engineered immunocompetent mice for heterozygous, conditional, epithelium-specific expression of the D305G caspase-8 mutation (D303 in humans) under the control of the endogenous caspase-8 promoter (Fig. 1A, Appendix Fig. 1). In The Cancer Genome Atlas (TCGA) PanCancer Atlas Studies (Cerami et al 2012; Gao et al 2013), mutation at the D303 site has been observed in 2 different human HNSCC tumors (D303G and D303V) and in a lung squamous cell carcinoma tumor (D303V). We have previously shown that the D303G and D303V caspase-8 mutant proteins retain the ability to homodimerize following stimulation of cells with the death ligand TRAIL but fail to mediate TRAIL-induced apoptosis (Cui et al 2021).
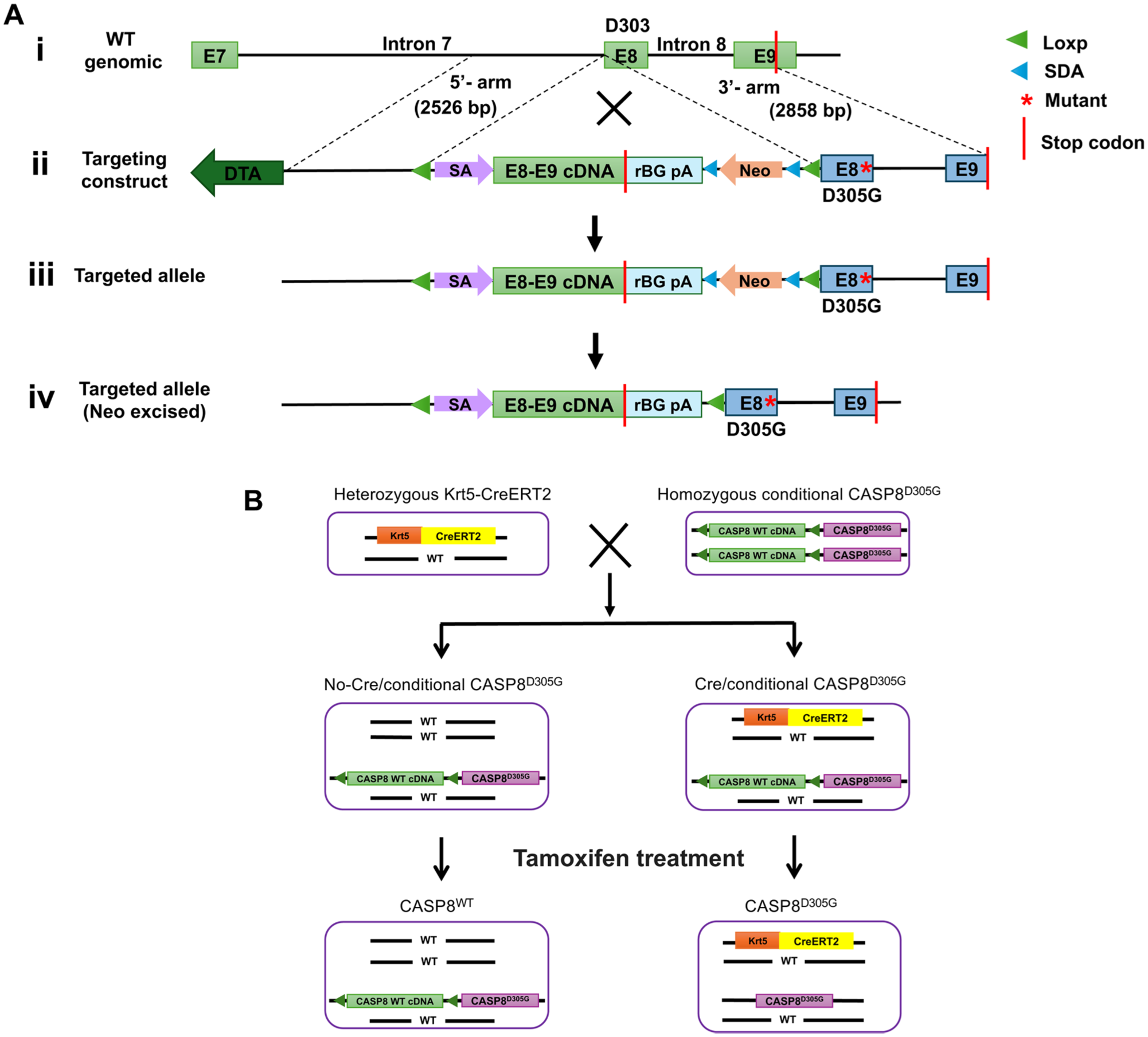
Generation of the caspase-8 D305G knock-in mice and tamoxifen treatment strategies. (
The targeting vector containing the D305G mutation is shown in Figure 1A. The D305 site is located in exon 8 of the murine caspase-8 gene and the stop codon in exon 9. The targeting vector contained a 5′ arm representing a portion of intron 7 (between exons 7 and 8). The 3′ arm of the targeting vector contained exon 8, into which the D305G mutation was introduced; intron 8 (between exons 8 and 9); and a portion of exon 9 containing the stop codon. Between the 5′ and 3′ arms, the construct contained a neomycin resistance gene flanked by self-deletion anchor (SDA) sequences, as well as the WT complementary DNA (cDNA) sequence encoding exons 8 and 9 (including the stop codon) flanked by loxP sites. The construct was designed so that in the absence of Cre, the construct would generate WT caspase-8, but following Cre-mediated excision of the WT cDNA sequence, the D305G mutant would be generated.
The targeting construct was introduced into C57BL/6 embryonic stem cells, and neomycin-resistant cells were injected into C57BL/6 blastocysts, followed by reimplantation into pseudo-pregnant female mice. The neomycin resistance gene was then removed by self-excision. Founder mice were genotyped for the presence of the desired targeting sequences, as described in the Materials and Methods, and Appendix Figure 1A, B. Founder mice that contained the targeting sequences were designated “conditional CASP8D305G” founders and were bred to homozygosity (Appendix Fig. 1C, D).
Homozygous, conditional CASP8D305G mice were then bred with heterozygous Krt5-CreERT2 mice (Fig. 1B, Appendix Figs. 2 and 3), in which CreERT2 expression is under the control of the keratin 5 promoter, enabling selective expression of tamoxifen-activated CreERT2 in epithelial tissues, including the oral epithelium. This breeding generated 2 lines of progeny mice (Fig. 1B). The first line of progeny mice lacked Krt5-CreERT2 sequences, were heterozygous for the conditional CASP8D305G sequence, and were designated “no-Cre/conditional CASP8D305G” (Fig. 1B, Appendix Figs. 2 and 3). Due to the lack of CreERT2 expression in these mice, treatment with tamoxifen did not result in excision of the loxP-flanked, WT caspase-8 exon 8/9 sequences and did not enable expression of mutant caspase-8 (Appendix Fig. 4A, C). Following tamoxifen treatment, these mice were referred to simply as “CASP8WT” mice (Fig. 1B). The second line of progeny mice, which were heterozygous for Krt5-ERT2 and heterozygous for the conditional CASP8D305G sequence, was designated “Cre/conditional CASP8D305G.” Tamoxifen treatment of “Cre/conditional CASP8D305G” led to excision of the WT caspase-8 exon 8/9 cDNA sequence and expression of the D305G caspase-8 mutant (Fig. 1B, Appendix Fig. 4B, C). Following tamoxifen treatment, these mice were referred to as “CASP8D305G” mice (Fig. 1B).
The D305G Caspase-8 Mutation Promotes Carcinogen-Induced Oral Tumor Formation
To determine the impact of caspase-8 mutation on HNSCC tumor development, CASP8WT and CASP8D305G mice were treated with the chemical carcinogen 4NQO (100 μg/mL in drinking water), and the formation, size, and pathological classification of resulting tumors were assessed. The study was initiated following a 2-wk resting period after tamoxifen treatment, with 31 CASP8WT mice and 24 CASP8D305G mice. Mice were given 4NQO for 16 wk, then returned to normal drinking water and examined for the development of tongue tumors 14, 28, and 41 d after the cessation of 4NQO treatment (Fig. 2A, B). No differences in body weights were detected following 4NQO treatment in the CASP8WT versus CASP8D305G groups (Appendix Fig. 5). For CASP8WT mice, the mean numbers of tongue tumors observed per mouse were 0.68, 0.81, and 1.3 on posttreatment days 14, 28, and 41, respectively (Fig. 2C, Appendix Table 1). In contrast, CASP8D305G mice exhibited means of 1.2, 1.3, and 2.9 tongue tumors per mouse on posttreatment days 14, 28, and 41, respectively (Fig. 2C, Appendix Table 1). Similarly, the percentage of mice with tongue tumors was markedly higher at each time point in the CASP8D305G group compared with the CASP8WT group (Fig. 2D, Appendix Table 1). However, although the mean numbers of tumors/mouse and the percentage of mice with tumors were higher in CASP8D305G mice, we did not observe a strong trend toward larger tumors in the CASP8D305G mice. Comparison of the CASP8D305G and CASP8WT groups 41 d after the end of 4NQO treatment revealed statistically significant differences between the groups only for smaller tumor sizes (<1 mm, 1–2 mm) and not for larger tumors (2–3 mm, >3 mm) (Fig. 2E, Appendix Tables 1 and 2).
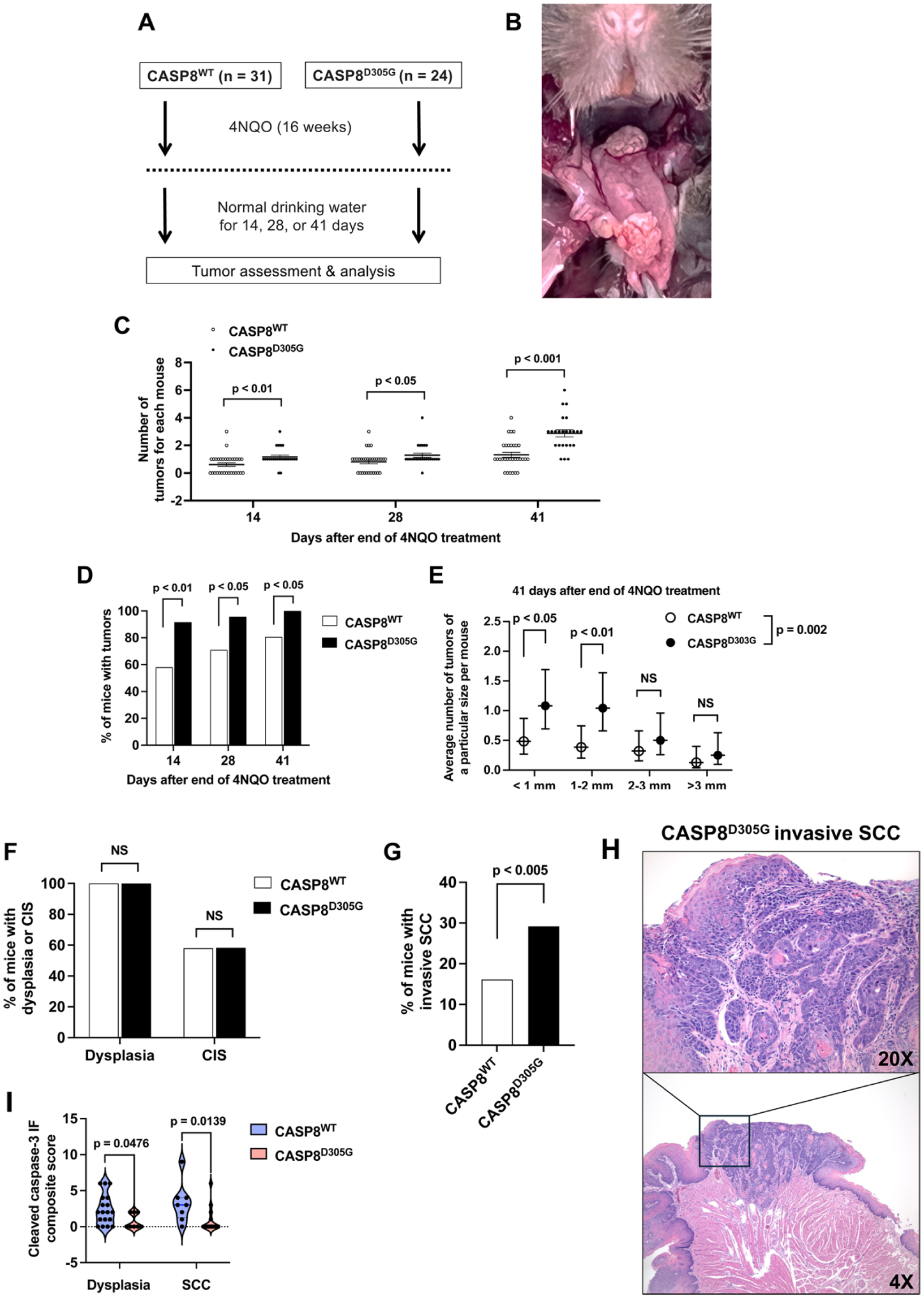
The caspase-8 D305G mutation promotes 4-nitroquinoline-1-oxide (4NQO)–induced development and progression of oral tumors. (
Caspase-8 Mutation Promotes Progression of Oral Tumors
We next performed hematoxylin and eosin (H&E) staining and pathology assessment of tumors harvested on day 41 after cessation of 4NQO treatment. At this time point, 80.7% of CASP8WT mice and 100% of CASP8D305G mice had observable tumors. H&E staining and pathology assessment were performed on the tongues from all mice, irrespective of the presence or absence of observable tumors. The percentage of mice with tongues exhibiting dysplasia (100% for both CASP8WT and CASP8D305G) or carcinoma in situ (58.1% for CASP8WT and 58.3% for CASP8D305G) did not differ between the CASP8WT and CASP8D305G groups (Fig. 2F, Appendix Table 3). By contrast, the percentage of mice with tongues exhibiting invasive squamous cell carcinoma was elevated in CASP8D305G mice (29.2%) versus CASP8WT mice (16.1%) (Fig. 2G, H, Appendix Table 3). Immunofluorescence staining of tumor tissues revealed reduced expression of cleaved caspase-3 in both dysplasia and squamous cell carcinoma from CASP8D305G mice compared with CASP8WT mice (Fig. 2I, Appendix Fig. 6). Taken together, our analyses of tongue tumor development and progression indicate that heterozygous mutation of caspase-8, as observed in human HNSCC, promotes carcinogen-induced oral tumor formation and progression in mice.
Caspase-8 Mutation Abrogates the Antitumor Activity of Anti–PD-1
The antitumor effects of T cells are mediated, in part, by activating caspase-8 in tumor cells. Therefore, we hypothesized that heterozygous expression of HNSCC-associated caspase-8 mutations in tumor cells would inhibit the antitumor activity of anti–PD-1. To test this, we used the MOC1 syngeneic tumor model. MOC1 cells were derived from a tongue tumor in carcinogen-treated C57BL/6 mice and grow well as tumors in these immunocompetent mice (Ku et al 2007; Judd et al 2012a, 2012b; Onken et al 2014). Blockade of PD-L1/PD-1 interactions has been reported to inhibit the growth of MOC1 tumors (Moore et al 2016a; Moore et al 2016b; Tran et al 2017). We first examined the impact of loss of caspase-8 on sensitivity to anti–PD-1 by comparing the response of tumors derived from parental (WT equivalent) MOC1 cells versus MOC1 caspase-8 knockout (MOC1 CASP8 KO) cells. Wild-type C57BL/6 mice were inoculated subcutaneously in the flank with parental MOC1 cells or MOC1 CASP8 KO cells. When tumors reached 100 mm3, mice in both groups were randomized to receive treatment with an isotype-matched control antibody or anti–PD-1 (0.2 mg/mouse). As shown in Figure 3A, treatment of tumor-bearing mice with anti–PD-1 resulted in significant growth inhibition of MOC1 parental tumors (P < 0.001) but not MOC1 CASP8 KO tumors, demonstrating the requirement for tumor expression of caspase-8 for the antitumor activity of anti–PD-1.
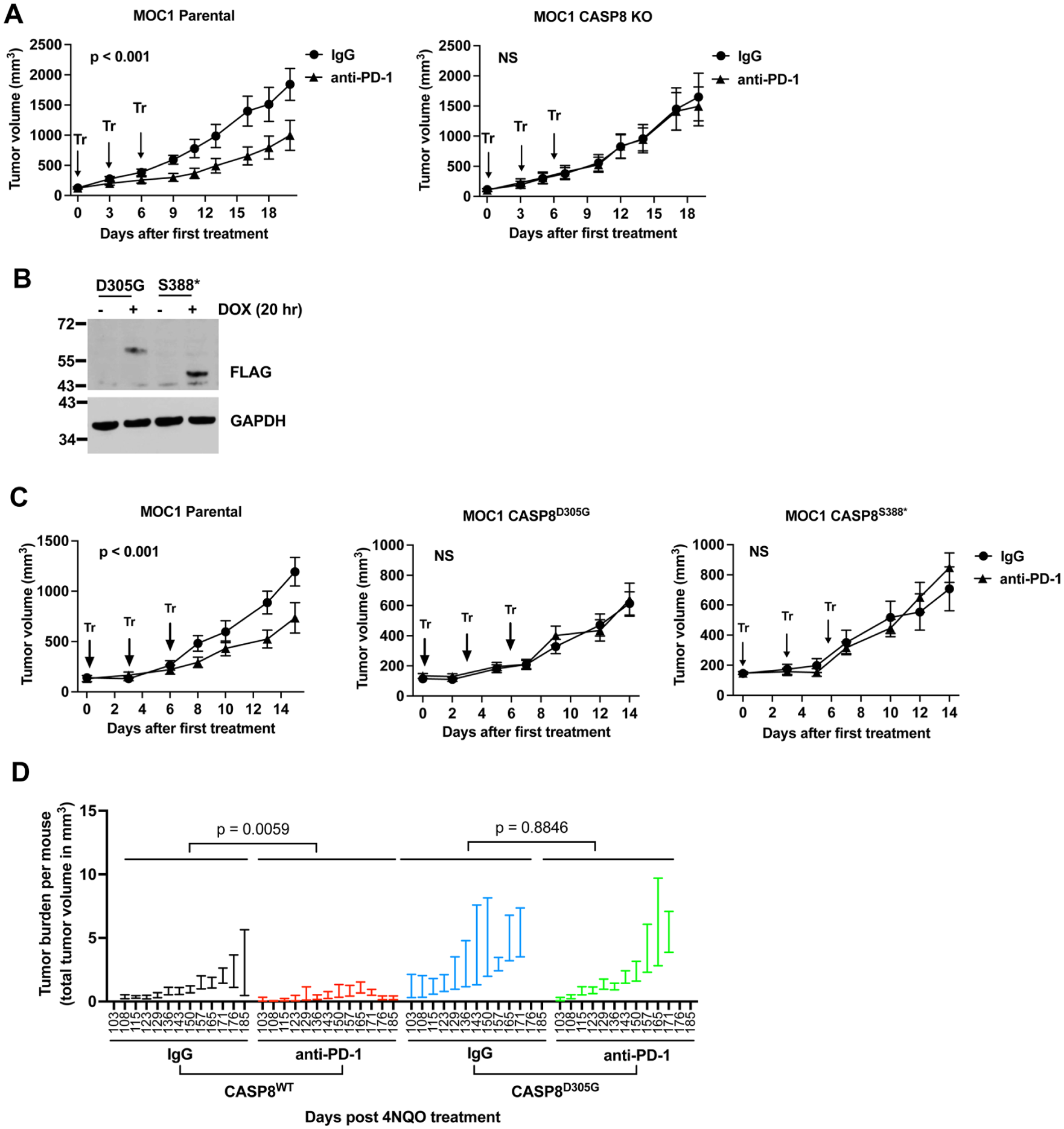
Heterozygous expression of caspase-8 mutants abrogates MOC1 tumor sensitivity to anti–PD-1. (
Because HNSCC-associated caspase-8 mutations are heterozygous in human tumors, we next examined the impact of heterozygous expression of caspase-8 mutants in MOC1 tumors on response to anti–PD-1. In addition to the D305G mutant, we also studied the effects of a second caspase-8 mutation, S388* (S386* in humans), observed in 2 different human HNSCC tumors in TCGA. MOC1 cells, which encode WT caspase-8, were engineered for doxycycline (DOX)–inducible expression of FLAG epitope-tagged D305G and S388* mutants, and DOX-induced expression of the mutant proteins was confirmed by immunoblotting with anti-FLAG (Fig. 3B). The cells were then inoculated into the flanks of C57BL/6 mice, and the mice were simultaneously given drinking water containing DOX (0.5 mg/mL) to induce expression of the mutant proteins. Treatment with isotype-matched IgG or anti–PD-1 was initiated when tumors reached 100 mm3. While MOC1 parental tumors again demonstrated significant growth inhibition in response to anti–PD-1, the MOC1 CASP8D305G and MOC1 CASP8S388* tumors failed to respond (Fig. 3C).
To exclude artifacts resulting from DOX treatment and the inducible system, we next generated MOC1 cells with constitutive, heterozygous expression of the caspase-8 D305G mutant using a T2A-mCherry cassette that drives separate expression of caspase-8 D305G and mCherry (Appendix Fig. 7A). Using the same treatment regimen, we observed that tumors constitutively expressing the caspase-8 D305G mutant were resistant to anti–PD-1, recapitulating our findings with the DOX-inducible system (Appendix Fig. 7B).
We next examined responsiveness to anti–PD-1 in 4NQO-induced tongue tumors in the CASP8WT and CASP8D305G mice. Tumor growth was quantified at each time point as total tumor burden per mouse. Consistent with our prior results, in this cohort, tumor burden developed earlier and was greater in CASP8D305G mice than in CASP8WT mice (Appendix Fig. 7C). Treatment with anti–PD-1 reduced tumor burden in CASP8WT mice but not in CASP8D305G mice (Fig. 3D). Collectively, these results demonstrate that tumors expressing HNSCC-associated caspase-8 mutations exhibit resistance to anti–PD-1 in immunocompetent mouse models.
Caspase-8 Mutation Modulates CD8+ T-Cell and Dendritic Cell Levels upon Anti–PD-1 Treatment
To further investigate how caspase-8 mutation may modulate response to anti–PD-1, we performed immunoprofiling. C57BL/6 mice were inoculated with MOC1 parental cells or MOC1 cells with constitutive, heterozygous expression of the caspase-8 D305G mutant. Fourteen days after inoculation, mice received a single dose of IgG or anti–PD-1 (0.2 mg/mouse), and tumors and spleens were harvested 36 h later for flow cytometric analysis (Fig. 4A).

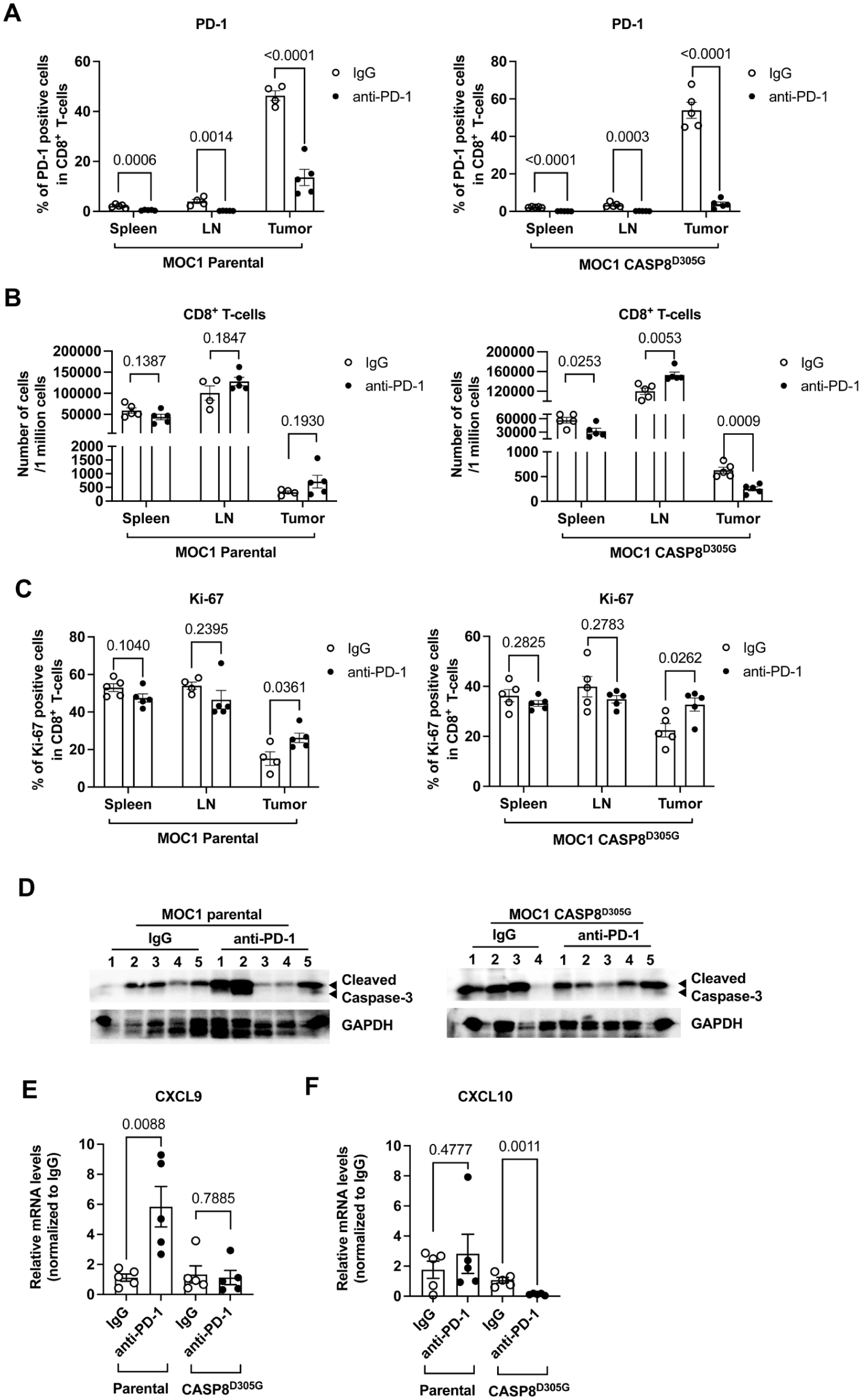
Caspase-8 mutation modulates CD8+ T-cell and dendritic cell (DC) levels after anti–PD-1. (
In mice bearing tumors derived from MOC1 parental cells, 1 dose of anti–PD-1 treatment did not significantly alter the intratumoral or splenic levels of total T cells (Appendix Fig. 8). By contrast, tumors with caspase-8 D305G exhibited a significant reduction in tumor-infiltrating and splenic total T cells following anti–PD-1 treatment, with a pronounced decrease in CD8+ T cells and their subsets (naive, central memory and effector CD8+ T cells), alongside reductions in natural killer (NK) and NKT cells (Fig. 4B, C). The levels of tumor-infiltrating CD4+ T cells were not impacted by anti–PD-1 treatment in both parental and mutant tumors (Appendix Fig. 9A), whereas splenic CD4+ T cells were significantly reduced in the caspase-8 mutant cohort, but not in the parental cohort, following anti–PD-1 treatment (Appendix Fig. 9B). B cells and Tregs were not significantly altered in tumors or spleens of both genotypes by anti–PD-1 versus IgG treatment (Appendix Fig. 10).
We further assessed the levels of myeloid lineage cells following treatment with anti–PD-1. In MOC1 parental tumors, anti–PD-1 increased intratumoral monocyte/dendritic cells but not granulocytes (Fig. 4D, Appendix Fig. 11A, B). Specifically, monocytes, total dendritic cells (DCs), and the conventional DC2 (cDC2, CD103–/CD11b+) subset, but not the monocytic myeloid-derived suppressor cells (mMDSCs) or tumor-associated macrophages (TAMs), were significantly elevated (Fig. 4D, Appendix Fig. 11C), with no corresponding changes in spleen (Appendix Fig. 11D), consistent with prior reports of monocyte and DC recruitment to tumors following PD-1 blockade (Schetters et al 2020; Lee et al 2021). In contrast, in caspase-8 mutant tumors, anti–PD-1 significantly reduced intratumoral monocytes, total DCs, and both cDC1 (CD103+/CD11b–) and cDC2 subsets relative to IgG, without changes in mMDSCs, TAMs, neutrophil, or granulocytic MDSCs (gMDSCs) (Fig. 4E, Appendix Fig. 11E). Most splenic myeloid populations were unchanged in mutant tumor-bearing mice, with only a slight increase in neutrophils observed following anti–PD-1 treatment (Appendix Fig. 11F). No significant differences in intratumoral M1/M2 ratio were observed across genotypes or treatments (Appendix Fig. 11G). Although the splenic M1/M2 ratio was increased after anti–PD-1 in both parental and caspase-8–mutant groups, these increases did not reach statistical significance (Appendix Fig. 11G). Collectively, these data suggest that tumor expression of the caspase-8 D305G mutant impairs the early intratumoral monocyte and DC response to PD-1 blockade and coincides with diminished CD8+ T-cell abundance locally and systemically.
Caspase-8 Mutation Disrupts Anti–PD-1–Induced CD8+ T-cell Redistribution and Chemokine Responses in Tumors
To further investigate how caspase-8 mutation affects the tissue distribution of immune cells after PD-1 blockade, we analyzed spleen, lymph node, and tumor samples from mice treated with anti–PD-1 or isotype IgG. PD-1 staining on CD8+ T cells was markedly reduced after anti–PD-1 treatment in all 3 tissues, consistent with receptor binding by the therapeutic antibody (Fig. 5A). In mutant tumor-bearing mice, anti–PD-1 induced a distinct tissue distribution of CD8+ T cells, characterized by reduced CD8+ T-cell abundance in tumors and spleens but increased CD8+ T-cell numbers in lymph nodes, a pattern not observed in parental tumors (Fig. 5B). Although anti–PD-1 increased the proportion of Ki-67+ CD8+ T cells within tumors in both parental and mutant groups, no increase was observed in spleens or lymph nodes (Fig. 5C), indicating that residual intratumoral CD8+ T cells in mutant tumors retain some proliferative responsiveness despite an overall reduction in tumor CD8+ T-cell abundance. IFN-γ increased only in splenic CD8+ T cells from mutant tumor-bearing mice after anti–PD-1 treatment, whereas TIM-3 decreased only in splenic CD8+ T cells from the mutant group (Appendix Fig. 12A, B). In contrast, LAG-3 and granzyme B were not significantly altered by genotype or treatment in any tissue examined (Appendix Fig. 12C). By comparison, monocyte and dendritic cell numbers were not significantly changed in spleens or lymph nodes after anti–PD-1 treatment (Appendix Fig. 12D, E). Anti–PD-1 increased cleaved caspase-3 in 3 of 5 tumors from parental mice but not in tumors from CASP8D305G mice, suggesting reduced tumor-cell apoptosis after PD-1 blockade (Fig. 5D). By contrast, phospho-MLKL was not detected in either group under these conditions, indicating that robust necroptosis was not induced by PD-1 blockade.
Caspase-8 mutation disrupts anti–PD-1–induced CD8+ T-cell redistribution, apoptotic engagement, and chemokine responses. (
To determine whether chemokine responses might contribute to the anti–PD-1 resistance of mutant tumors, we measured tumor chemokine expression after treatment with IgG or anti–PD-1. In parental tumors, anti–PD-1 increased CXCL9 expression, whereas this induction was absent in caspase-8–mutant tumors (Fig. 5E). In addition, CXCL10 expression decreased in anti–PD-1–treated caspase-8–mutant tumors but not in parental tumors (Fig. 5F), while CCL2 and CCL5 remained unchanged (Appendix Fig. 13). Together, these findings suggest that caspase-8 mutation impairs anti–PD-1–induced local immune remodeling, characterized by blunted CXCL9/CXCL10 chemokine response, thereby limiting the transition of CD8+ T cells from lymph nodes to tumors.
Discussion
Genomic studies have revealed that the gene encoding the apoptotic protease caspase-8 is among the more frequently mutated genes in HNSCC. Mutations in the caspase-8 gene are dispersed throughout the coding region, with the lack of hotspot mutations suggesting that the caspase-8 protein may have tumor suppressor activity. Further, heterozygous expression of HNSCC-associated caspase-8 mutations has been shown to block, in a dominant-negative fashion, death ligand–induced apoptosis (Li et al 2014; Cui et al 2021). Our studies now reveal that heterozygous expression of a caspase-8 mutation under the control of the endogenous caspase-8 gene promoter stimulates HNSCC tumor development in vivo in immunocompetent mice, clearly implicating the importance of the mutations.
Our studies also provide important insight into a possible mechanism of resistance to ICI therapy in HNSCC. Because effector T cells exert their antitumor effects, in part, by activating cell death induced by death ligands, we examined whether heterozygous caspase-8 mutations would impact the in vivo response of HNSCC tumors to anti–PD-1. Our findings show that the antitumor activity of PD-1–targeting antibody is blocked when tumor cells express heterozygous caspase-8 mutations. This suggests the need to evaluate whether caspase-8 mutation is associated with failure to respond to immunotherapy in the subset of patients with HNSCC whose tumors harbor such mutations.
Unexpectedly, in mice bearing caspase-8 mutant tumors, anti–PD-1 treatment reduced CD8+ T-cell levels in both tumor and spleen, as well as diminished DC and monocyte levels in tumors. PD-1 blockade typically results in rapid TNF-α production from functionally restored T cells or other innate immune cells (Zhou et al 2010; Simon et al 2016; Howard et al 2021). Our immune profiling analysis in the draining lymph node suggests this phenotype aligns more with impaired local immune-cell recruitment within the mutant tumors than with a broad defect in immune activation. The absence of CXCL9 induction and reduction of CXCL10 in caspase-8 mutant tumors after anti–PD-1 treatment further supports this interpretation. In addition, the reduced expression of cleaved caspase-3 in caspase-8–mutant tumors likely reflects both reduced local immune-cell accumulation and reduced cell susceptibility to apoptosis. However, the precise mechanisms by which caspase-8 mutations modulate these chemokine responses after anti–PD-1 treatment and how this affects the redistribution of DCs and T cells require further investigation.
At present, there is no publicly available HNSCC dataset that integrates caspase-8 mutation status with clinical response to ICIs. Examination of TCGA HNSCC data reveals that CASP8-mutant tumors do not uniformly exhibit an immune-excluded phenotype, and in some HPV-negative HNSCC cases, mutation is associated with higher immune infiltration and stronger interferon-related signatures despite poorer clinical outcomes. This apparent difference from our mouse models likely reflects the biological heterogeneity of human HNSCC tumors, including heterogeneity of co-occurring genetic alterations, and underscores the need for future studies integrating tumor genetics with immunotherapy response in HNSCC clinical cohorts.
In summary, our preclinical results strongly support clinical investigation to determine whether the caspase-8 mutation is associated with failure to respond to ICI therapy in patients with HNSCC. Moreover, as pembrolizumab and/or nivolumab have been approved by the US Food and Drug Administration in several other cancers where caspase-8 mutations are prevalent (endometrial, stomach, cervical, colorectal, and bladder cancers and melanoma), the impact of this potential mechanism of resistance may be broad.
Author Contributions
Z. Cui, H. Wu, contributed to conception and design, data analysis, drafted and critically revised the manuscript; L.C. Woerner, S.R. Long, C.N. Peterson, N.K. VanLandingham, A. Nazarenko, M.-O. Kim, J.R. Grandis, D.E. Johnson, contributed to data analysis, critically revised the manuscript. All authors gave their final approval and agree to be accountable for all aspects of the work.
Supplemental Material
sj-docx-1-jdr-10.1177_00220345261457066 – Supplemental material for Caspase-8 Mutation Promotes HNSCC Development and Resistance to Anti–PD-1
Supplemental material, sj-docx-1-jdr-10.1177_00220345261457066 for Caspase-8 Mutation Promotes HNSCC Development and Resistance to Anti–PD-1 by Z. Cui, H. Wu, L.C. Woerner, S.R. Long, C.N. Peterson, N.K. VanLandingham, A. Nazarenko, M.-O. Kim, J.R. Grandis and D.E. Johnson in Journal of Dental Research
Supplemental Material
sj-pdf-2-jdr-10.1177_00220345261457066 – Supplemental material for Caspase-8 Mutation Promotes HNSCC Development and Resistance to Anti–PD-1
Supplemental material, sj-pdf-2-jdr-10.1177_00220345261457066 for Caspase-8 Mutation Promotes HNSCC Development and Resistance to Anti–PD-1 by Z. Cui, H. Wu, L.C. Woerner, S.R. Long, C.N. Peterson, N.K. VanLandingham, A. Nazarenko, M.-O. Kim, J.R. Grandis and D.E. Johnson in Journal of Dental Research
Footnotes
Acknowledgements
Cyagen US generated the tamoxifen-inducible CASP8D305G knock-in mice. The authors thank the Core Animal Facility of the Helen Diller Family Comprehensive Cancer Center for support and advice with mouse breeding and analysis. Flow cytometric data in this study were acquired at the Optical Imaging and Analysis Facility, School of Dental Medicine, State University of New York at Buffalo.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: J.R.G. and D.E.J. are coinventors of a cyclic STAT3 decoy oligonucleotide and report financial interests in Bluedot Bio LLC, the exclusive licensee of the cyclic STAT3 decoy patents.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institutes of Health grants R01DE024728 (D.E.J.) and R35CA231998 (J.R.G.) and by a California Tobacco-Related Disease Research Program award (T29FT0328) and New York State Research Foundation to Z.C.
Ethics Approval and Consent to Participate
This study did not require ethics approval or consent to participate because no human subjects, human patient data, or human-derived materials were used. All experiments were performed with established cell lines or animal models. Ethics approval for studies using mice was obtained from the University of California–San Francisco Institutional Animal Care and Use Committee (IACUC protocol #AN197429-01C), and all experiments were performed in accordance with established guidelines.
Data Availability
All data were generated by the authors and are included in the article, and no other public data were used in this study.
A supplemental appendix to this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.