
Abstract
Objective
Spasmodic dysphonia is a focal dystonia of the larynx with heterogeneous manifestations and association with familial risk factors. There are scarce data to allow precise understanding of etiology and pathophysiology. Screening for dystonia-causing genetic mutations has the potential to allow accurate diagnosis, inform about genotype-phenotype correlations, and allow a better understanding of mechanisms of disease.
Study Design
Cross-sectional study.
Setting
Tertiary academic medical center.
Subjects and Methods
We enrolled patients presenting with spasmodic dysphonia to the voice clinic of our academic medical center. Data included demographics, clinical features, family history, and treatments administered. The following genes with disease-causing mutations previously associated with spasmodic dysphonia were screened: TOR1A (DYT1), TUBB4 (DYT4), and THAP1 (DYT6).
Results
Eighty-six patients were recruited, comprising 77% females and 23% males. A definite family history of neurologic disorder was present in 15% (13 of 86). Average age (± standard deviation) of symptom onset was 42.1 ± 15.7 years. Most (99%; 85 of 86) were treated with botulinum toxin, and 12% (11 of 86) received oral medications. Genetic screening was negative in all patients for the GAG deletion in TOR1A (DYT1) and in the 5 exons currently associated with disease-causing mutations in TUBB4 (DYT4). Two patients tested positive for novel/rare variants in THAP1 (DYT6).
Conclusion
Genetic screening targeted at currently known disease-causing mutations in TOR1A, THAP1, and TUBB4 appears to have low diagnostic yield in sporadic spasmodic dysphonia. In our cohort, only 2 patients tested positive for novel/rare variants in THAP1. Clinicians should make use of genetic testing judiciously and in cost-effective ways.
Keywords
Spasmodic dysphonia (SD) is a chronic progressive neurologic disorder of central motor processing, with action-induced, task-specific focal laryngeal dystonia and no cure available. 1 Females are preferentially affected, and the average age of symptom onset is 45 years. 2 Some cases can be highly disabling with significant psychosocial impact.3,4 Variants are classified according to the predominant vocal cord muscles affected, including adductor (most prevalent), abductor, and mixed types. The condition may coexist with vocal tremor as well as other nonvocal tremors. 5 Current treatments are largely targeted at symptomatic improvement, including recurrent sessions of chemodenervation of laryngeal muscles with botulinum toxin and denervation-reinnervation surgery on select cases.6,7
There is little information regarding the pathogenesis of SD. Although functional, neurochemical, and neuropathologic abnormalities have been found, data are scarce to inform a comprehensive disease model.8-10 In recent years, a growing interest has emerged in studying genetic contributions to focal dystonias, stemming from evidence that polymorphisms in wild-type forms of generalized dystonia-causing genes can affect the risk of developing focal or segmental dystonia as well as the clustering of heterogeneous manifestations of dystonia in some families.11-13
Previous genetic variants that have been studied in association with focal dystonia comprising SD include DYT1/TOR1A12,14 and DYT6/THAP1.15-17 Results are either conflicting or have not been reproduced. More recently, mutations in the DYT4/TUBB4 gene were identified in an Australian family, with the phenotype of whispering dysphonia originally described by Parker and more recently reappraised by Wilcox et al.18-21 Studies analyzing the prevalence of pathogenic variants in coding regions of TUBB4 in primary dystonia have not identified pathogenic variants in large-scale cohorts. However, patient selection in these studies was not restricted to SD phenotypes.22,23 To our knowledge, this is the first report to exclusively screen patients with SD, describing epidemiologic data and family history, and performing analysis of genetic abnormalities in coding regions of TOR1A, THAP1, and TUBB4.
Methods
Subjects with SD were recruited from the Voice Center Clinic at the Massachusetts Eye and Ear Infirmary. Participants signed informed consent as approved by our Institutional Review Board. A physical examination was followed by speech recordings, which were independently scored regarding the presence of at least 1 voice break in a vowel per 3 of 10 sentences useful for eliciting glottal stops in vowels for identifying adductor SD, prolonged voiceless consonants in sentences for identifying abductor SD, and prolonged vowels for identifying vocal tremor. A speech language pathologist then evaluated each subject. The evaluation, including the flexible digital nasolaryngoscopic examination, was digitally recorded for subsequent review. The larynx was evaluated during quiet breathing, quiet sustained /i/, and loud sustained /i/ vocalization, followed by repeating some of the sentences that the patient recited earlier for the speech recording. The presence and the degree of symmetry of vocal fold adduction or abduction and ventricular hyperfunction were recorded during whistling, voiced speech, and whispered speech. The history, physical examination, and nasolaryngoscopic examination were reviewed at weekly multidisciplinary voice rounds. During this session, a panel of 2 experienced laryngologists (P.C.S., R.A.F.) and 6 speech language pathologists reviewed the findings. Each panel member rated the following parameters—on a visual analog scale (0-100 mm) ranging from normal to asymmetric—for the following qualities: anatomic symmetry while breathing, functional symmetry while breathing, tremor at rest, tremor during voice (prolonged vowel), spasms during vocalizing sentences (the 10 sentences used above), and when repeating specific syllables (si-si-si-, I-I-I, hi-hi-hi). All members of the panel then reached a consensus regarding diagnosis. A final diagnosis was made of adductor SD, abductor SD, vocal tremor, muscular tension dysphonia, or “other.” Only those subjects with a consensus diagnosis of adductor SD or abductor SD were eligible to participate in this study. 24
Subsequently, all subjects were examined by a movement disorder neurologist (N.S.) to confirm the diagnosis of SD and determine if there was evidence of dystonia elsewhere, according to published criteria and revised according to a recent update.25,26 Subjects were asked about any associated events that preceded the onset of SD, such as viral illness, trauma, or stress (eg, divorce, marriage, loss of vocation, relocation to a new city). The presence of a family history of dystonia was determined, for first- and second-degree relatives, as previously described. 27 Subjects were considered to have a positive family history of dystonia if the first- or second-degree relative with dystonia provided medical records confirming the diagnosis or was examined by one of the clinical investigators who participated in this study (N.S.). Subjects were considered to have a possible family history if they reported affected first- or second-degree members but confirmatory medical records or examination were not obtained. A negative family history was recorded if subjects specifically denied such a history. The local Institutional Review Board approved the study, and all participating individuals gave informed consent.
Molecular Analysis
DNA was extracted from white blood cells through the Purgene procedure (Gentra Systems, Minneapolis, Minnesota).
The samples were screened for presence of the TOR1A (DYT1) GAG deletion as well as THAP1 (DYT6) and TUBB4A (DYT4) mutations. Polymerase chain reaction amplification across the GAG deletion region of the TOR1A gene was performed as previously described.27,28 All exons and flanking regions of the THAP1 gene and the TUBB4A gene were sequenced as previously described.17,20 Variants were compared with frequencies reported in the Exome Aggregation Consortium (ExAC) database. 28
Results
A total of 87 subjects were enrolled in the study. One patient declined genetic screening and was excluded from the final analysis. Clinical and demographical data can be seen on Table 1 . Average age (± standard deviation) of symptom onset was 41.1 ± 16.4 years, the majority with late onset (defined as ≥27 years; 84%, 72 of 86) and a female predominance (77%; 66 of 86). All subjects had onset of dystonia in the larynx, and in the majority it remained focally restricted to the vocal folds (84%, 73 of 86; Figure 1 ). Data regarding the type of SD were available in 85 subjects. The majority had adductor SD (89%, 76 of 85), with smaller subsets having abductor (5%, 5 of 85) or mixed (4%, 4 of 85) type. There was segmental spread in 10% of subjects and multifocal involvement in 5%. Seven subjects (8%) had tremor. Of these, 5 exhibited a postural upper extremity tremor (2 in combination with head/neck and voice tremor), and 2 had isolated head/neck tremor. Forty-one percent (35 of 86) of subjects associated onset with ≥1 risk factors—most commonly, psychosocial stressors, frequent voice use, upper respiratory infections, or trauma ( Figure 2 ).
Demographic and Clinical Data (N = 86).
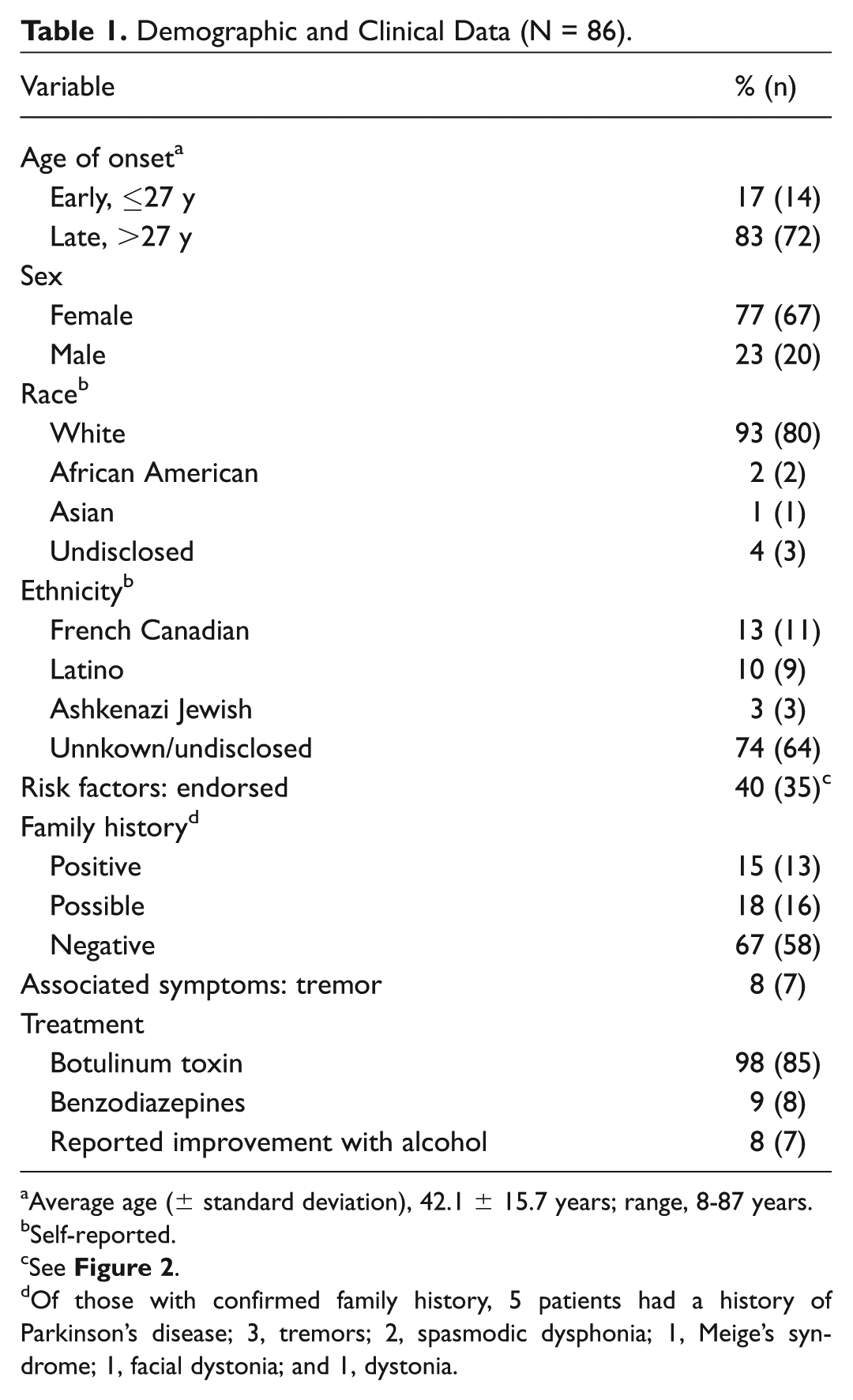
Average age (± standard deviation), 42.1 ± 15.7 years; range, 8-87 years.
Self-reported.
See Figure 2 .
Of those with confirmed family history, 5 patients had a history of Parkinson’s disease; 3, tremors; 2, spasmodic dysphonia; 1, Meige’s syndrome; 1, facial dystonia; and 1, dystonia.
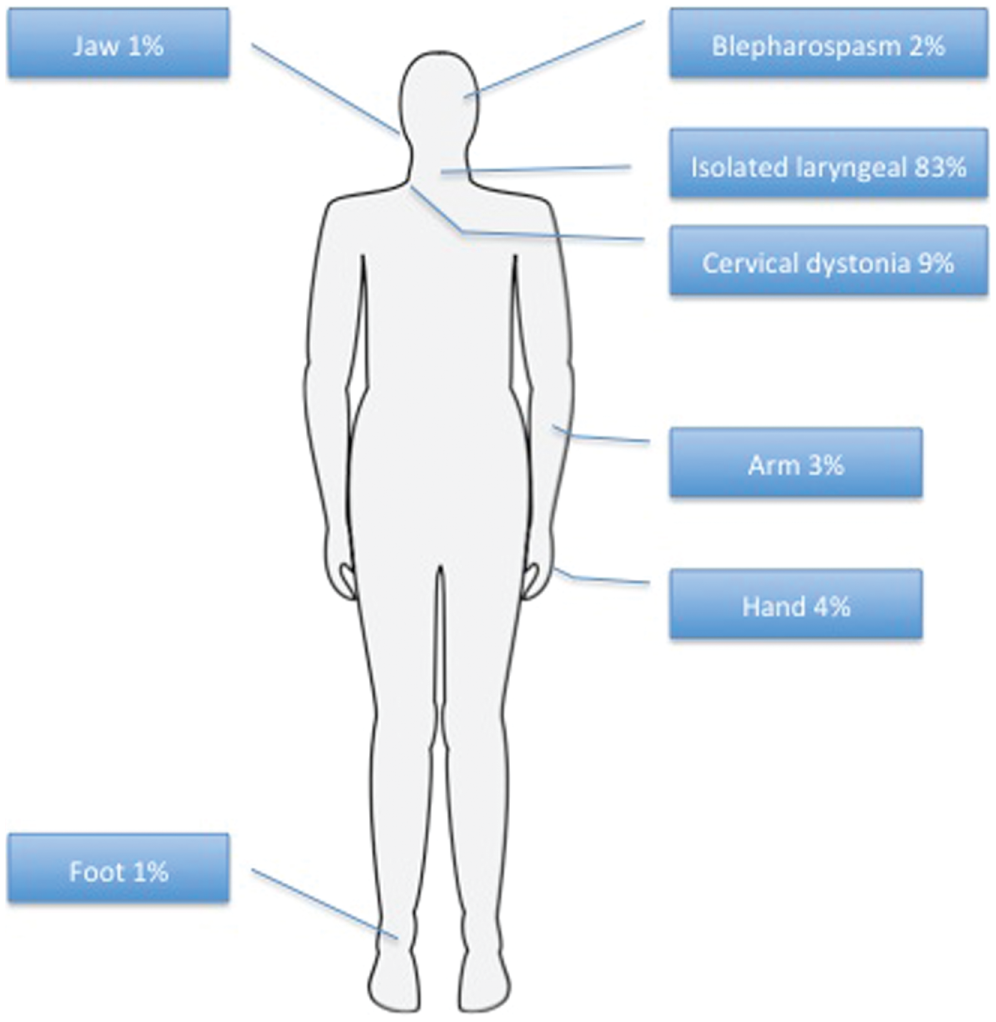
Frequency of dystonia distribution in spasmodic dysphonia subjects.
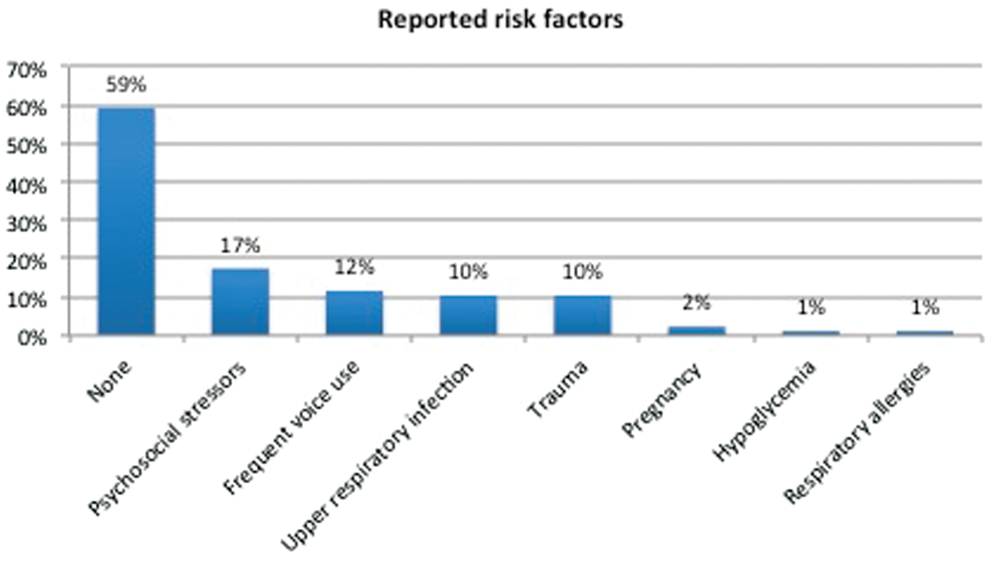
Frequency of reported risk factors preceding onset of spasmodic dysphonia.
Thirteen subjects (15%) had a definite family history of neurologic disorder, with 5 specifically having a first-degree family member with dystonia. Other subjects with a positive family history (first or second degree) included Parkinson’s disease (n = 5) and essential tremor (n = 3).
Most subjects were treated with electromyography-guided botulinum toxin (98%; 85 of 86). One patient declined injections. A fraction of patients (11 of 86, 13%) received oral medications, most commonly benzodiazepines. All patients treated with botulinum toxin had a positive response, confirming the diagnosis of SD.
Genetic screening was negative in all patients for the GAG deletion in TOR1A (DYT1) and for disease-causing mutations in TUBB4A (DYT4). Sequence analysis in 2 patients revealed potential mutations in the THAP1 gene. In a white male patient of unknown descent with an early onset (age, 24 years) and no family history, a novel c.11C>T (Chr8:42,698,227;hg19) change was identified, resulting in a p.Ser4Phe amino acid substitution. This variant was not found in ExAC, dbSNP, ClinVar, or any other variant database and was predicted to be deleterious by SIFT and possibly damaging by PolyPhen. This patient enrolled in the study at age 70 years and thus had not had any progression in decades, remaining well controlled with botulinum toxin injections. A female patient, with a history of Graves’ disease, had onset of focal SD at age 50 years. She reported a positive family history of Meige’s syndrome in her mother. She had Italian, Portuguese, and Irish ancestry. In 9 years of follow-up, she has been stable with periodic botulinum toxin injections and no signs of progression. This patient harbored a c.580T>C (chr8:42,693,167;hg19) missense variant resulting in a p.Ser194Pro substitution. This variant has been reported in an individual of East Asian ancestry in the ExAC data set and is predicted to be tolerated by SIFT and benign by PolyPhen, suggesting that this may be a rare benign variant or a variant of unknown significance in this patient. Unfortunately, a DNA sample from the mother is not available.
Discussion
In this sample of 86 patients with mainly sporadic SD, genetic screening for disease-causing mutations in known dystonia genes previously implicated in SD (TOR1A, THAP1, and TUBB4A) identified 1 mutation and 1 variant of unknown significance in the THAP1 gene. In terms of patient population, age of onset, and reported risk factors, this cohort is consistent with previous epidemiologic data.1,29,30
Our findings suggest a low diagnostic yield for indiscriminate genetic screening in a nonselected population of SD patients. The absence of mutations in the TOR1A and TUBB4 is informative. TOR1A is classically described as having onset in a limb during late childhood, with generalization typically sparing the craniofacial muscles, but variations have been reported.31,32 TUBB4 mutations have been associated with isolated SD to generalized dystonia with a characteristic gait and dysphonia. These phenotypes appear to be restricted to the original kindred, although leukoencephalopathies with hypomyelination are increasingly recognized in association with particular mutations in this gene, putatively related to a continuum of phenotypic expressivity.18,19,22,33,34
The presence of novel/rare variants in THAP1 in 2 of our patients is of interest, but the conclusions are limited. THAP1 mutations appear to be present but rare in focal and segmental SD patients. 17 Previous studies examining genotype-phenotype correlations suggest that computationally predicted pathogenic mutations or those affecting the DNA-binding domain most often manifest with younger age of onset and more severe disease (generalized dystonia).35,36 Nevertheless, our patient with the p.Ser4Phe missense mutation in the DNA-binding domain had relatively early onset of disease (age, 24 years) but has remained restricted to the larynx for several decades.
There are limitations to our study. Our sample size is relatively small despite its representativeness of typical SD. Previously published data report similar age of onset, female sex predominance, environmental risk factors, and response to treatment.1,2,29,30 In addition, we restricted the evaluation to GAG deletions in the coding region of TOR1A. Other disease-causing mutations in this gene have been described but appear exceedingly rare. As previously mentioned, there may be higher yield in testing those with early-onset speech involvement associated with progression to generalized dystonia. 17 In this cohort, 16% (14 of 86) of patients had early onset, restricted to the larynx in 78% (11 of 14). In 1 patient with early onset (7%; 1 of 14) a pathogenic THAP1 mutation was found. This finding is in line with the aforementioned studies but also unique, as the reported patient did not have progression to generalized dystonia.
In summary, our findings suggest that genetic screening for mutations in DYT1/TOR1A, DYT6/THAP1, and DYT4/TUBB4A in patients with sporadic SD has low diagnostic yield and should not be routinely performed. Targeted screening is presently more likely to have higher yield in those with generalized dystonia, young onset, and a positive family history. With widespread use of clinical exome/whole genome sequencing, knowledge of phenotypic expression of monogenic conditions is likely to expand. In this scenario, candidate gene screening will probably need to be revisited in the near future.
Author Contributions
Disclosures
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
This article was presented with preliminary data at the American Academy of Neurology Meeting; April 2015; Washington, DC.