
Abstract
Hypertrophic cardiomyopathy (HCM) characterized by asymmetric ventricular septal hypertrophy, is the commonest cause of sudden cardiac death (SCD) in the young. The underlying etiology of HCM in the childhood and adolescent patients is diverse. Moreover, the prognosis of pediatric HCM depends on the age of presentation and etiology. Despite the complexity of children with obstructive HCM, surgical treatment results in a favorable outcome for carefully selected patients in experienced tertiary referral center in contemporary era. Implantable cardioverter-defibrillator (ICD) remains the most effective and reliable treatment to prevent SCD. New pediatric SCD risk prediction model, which has good discrimination and calibration and can distinguish patients who are most benefit from an ICD implantation, is expected to be further refined in the future.
Introduction
Hypertrophic cardiomyopathy (HCM) characterized by asymmetric ventricular septal hypertrophy, is the most common monogenic myocardial disease and the commonest cause of sudden cardiac death (SCD) in the young.1–4 HCM perhaps differs largely from other cardiovascular diseases in the view of clinical presentation and natural course, due to its incomplete penetrance and variable expressivity.5,6 The clinical manifestation of HCM may present at any age from infancy to advanced age and vary from no symptom to severe symptom, or even SCD. This review focuses on HCM in children and aims to provide an overview of this highly specialized patient population.
Epidemiology
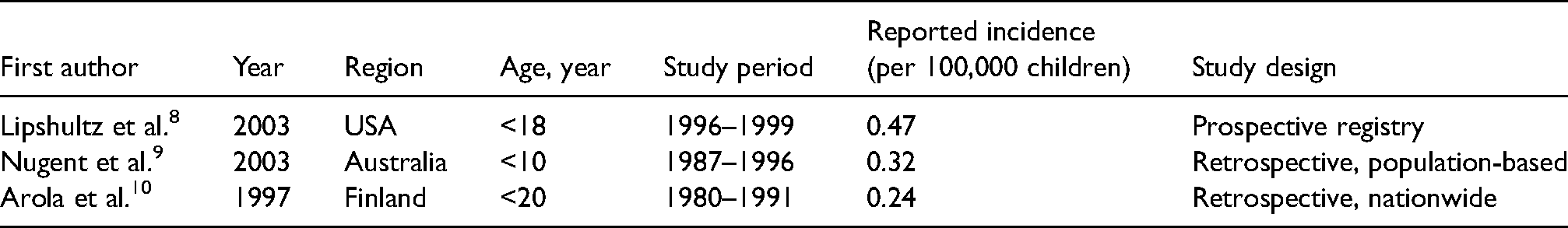
Although HCM is a relatively common genetic cardiovascular disorder in the adult population, children with HCM are rare. To date, most of the available information concerning HCM comes from studies conducted in adult populations, which is evidenced by the consistently reported prevalence of 1 in 500 adults. 7 On the contrary, the prevalence of HCM in children remains unknown. Only a handful of epidemiological studies limited to North America and Europe have investigated the annual incidence of HCM in children (Table 1).8–11 The Nationwide Study in Finland carried out the first well-defined population-based retrospective study after the development of modern echocardiographic techniques, to determine the epidemiology of idiopathic cardiomyopathies in children, including HCM. 11 During the 12-year study period, the estimated annual incidence of HCM in children was 0.24 per 100,000 children below 20 years of age. In 2003, the National Australian Childhood Cardiomyopathy Study and the Pediatric Cardiomyopathy Registry of the United States presented an annual incidence of HCM in children of 0.32 per 100,000 children below 10 years of age and 0.47 per 100,000 children below 18 years of age, respectively.8,9
Reported incidences of paediatric hypertrophic cardiomyopathy.
Etiology and genetics
HCM which is defined as morphological hypertrophy of the left ventricle and generally used in current clinical practice, is actually a purely descriptive nomenclature regardless of cause.12,13 The underlying etiology of HCM is diverse, particularly in childhood and adolescent patients.4,13 However, unlike that of adult HCM, population-based observational studies investigating the underlying cause of HCM in children are currently sparse.2,4 Recently, a retrospective, longitudinal multi-centre cohort study of paediatric HCM in the UK reported that non-syndromic cause accounted for 63% of the underlying etiology. 14 Nevertheless, only 23% of pathogenic mutations were cardiac sarcomere protein genes in this study. The aforementioned study is the largest study from Europe, which described a cohort of patients under 16 years of age. Before that, the current understanding of the etiology of childhood HCM is primarily derived from the North American registry, which enrolled 855 patients under 18 years of age and is the largest study in paediatric HCM up to now. 13 Results of the Pediatric Cardiomyopathy Register from Canada and the US classified 74.2% of cases as idiopathic HCM, which was defined as patients without known cause. 13 Results of both studies are in agreement with that the underlying etiology of paediatric HCM is heterogeneous. Conversely, the National Australian Childhood Cardiomyopathy Study identified a syndromic, metabolic, or genetic explanation in nearly 60% of subjects, including Noonan syndrome in 28.8%. 4 Of note, the latter only included 80 children with HCM under 10 years of age and did not present detailed results of genetic testing.
Clinical course
Limited available data suggest that the prognosis of paediatric HCM depends on the age of presentation and etiology.3,4,13–17 The National Australian Childhood Cardiomyopathy Study is the first population-based cohort study, which includes all children in Australia presenting with primary cardiomyopathy at 0 to 10 years of age between 1 January 1987 and 31 December 1996 and in which a total of 80 subjects with HCM were identified. 4 In this study, the average annual mortality for the entire study population was 3.39% and 1.52% for subjects diagnosed after the first year of life during the follow-up period. Furthermore, for each of those identified risk factors, the highest mortality was during the first 12 months after presentation. Additionally, the 5-year survival for children diagnosed during the first year of life was worse compared with that for those diagnosed beyond 1 year of age. At last, the authors concluded that prognosis among children with HCM is better than that previously reported by institutional reviews. In the year of 2007, the Pediatric Cardiomyopathy Registry reports the outcome of HCM in children, including its primary (isolated) and secondary (systemic) forms. 13 This registry study enrolled 855 children with pure HCM defined as a normal systolic ventricular function, which comprised a prospective cohort and a retrospective cohort. The main findings are consistent with that observed in the National Australian Childhood Cardiomyopathy Study, though the Pediatric Cardiomyopathy Registry contained a much larger patient population in a wider age range.13,17 The presence of multiple risk factors at diagnosis indicated the worst outcomes in most etiologic subgroups. 17 Not only the absolute risk of death or heart transplantation significantly increases in the presence of multiple risk factors, but also the risk increases significantly as the number of risk factors increases. 17
Surgical treatment
Surgical technique
As described previously, an extended septal myectomy was performed, which evolved from the classic Morrow procedure. 18 Via a transverse aortotomy, aortic valve commissures were suspended with pledgeted sutures to expose the hypertrophied septum. The first longitudinal incision was initiated at 4–5 mm below the middle of the right coronary cusp, and then extended distally to the left ventricular apex. The second parallel incision initiated at 4–5 mm below the commissure of the right and left cusps, and then extended to the level of the mitral anterior commissure. The third horizontal incision below the aortic annulus connected former two incisions. These three incisions were accomplished by using the 12th scalpel blade, which resulted in a trough on the anterior interventricular septum. Then the intact specimen was removed from the interventricular septum by using the 15th round blade as far as possible. Besides, right ventricular (RV) outflow tract (RVOT) obstruction which was defined with the peak RVOT pressure gradient of 30 mm Hg or more and intraventricular anatomical abnormalities in the left ventricular including but not limited to abnormal muscular bundles, fusion of papillary muscles, and anomalous chordae, were examined carefully and manipulated accordingly as previously described. 19 Concomitant RV obstruction relief, including septal myectomy, infundibular resection, RVOT patch enlargement with an autologous pericardial or Dacron patch, or a combination of them, was performed via subpulmonary longitudinal ventriculotomy. Concomitant surgery was performed according to corresponding indication or expert consensus. If intraoperative transesophageal echocardiography showed the presence of residual gradient higher than 30 mm Hg or persistent systolic anterior motion of the anterior mitral leaflet, cardiopulmonary bypass would be resumed to perform additional resection. A temporary pacemaker was routinely placed immediately following septal myectomy.
Surgical result
Surgical septal myectomy is the gold standard treatment for adult HCM patients with left ventricular outflow tract obstruction and severe drug-refractory symptoms.12,20,21 Multiple institutional studies from dedicated HCM centres have reported sustained improvement of survival, exercise capacity and quality of life.18,22–26 However, available data on surgical treatment in children with obstructive HCM are sparse, which are limited to either small samples or mixed populations not exclusively including children under 18 years old.19,27–31
Up to December 2020, we have performed myectomy in 160 children (Figure 1). We have recently reviewed our experience of 117 consecutive children with obstructive HCM, who were aged 0.6 to 17.5 years and operated between February 2009 and December 2018. 19 Our study sought to describe the overall phenotypic characteristics of children with obstructive HCM and relevant surgical outcomes. There were 22 patients (18.8%) presenting with RV outflow tract obstruction simultaneously. In addition, 25 patients (21.4%) had myocardial bridging of at least one coronary artery. Besides, left ventricular anatomic abnormalities were observed in 61 patients (52.1%) (Figure 2). After surgery, one 1.6-year-old boy (0.9%) died of heart failure and subsequent multi-organ failure. Representative specimen removed from hypertrophied septum was presented in Figure 3. During follow-up, 3 patients (2.6%) died suddenly at 1.3, 1.5, and 2.1 years after surgery and were all less than 18 years of age at the time of death, yielding overall survival rates of 100% at 1 year and 96.5% at 3 years. Only 2 patients (1.7%) required cardiac reoperation, but neither underwent repeat myectomy, yielding overall reoperation-free survival rates of 99.1% at 1 year and 98% at 3 years. Despite the complexity of children with obstructive HCM, surgical treatment results in a favourable outcome for carefully selected patients in an experienced tertiary referral centre in the contemporary era.
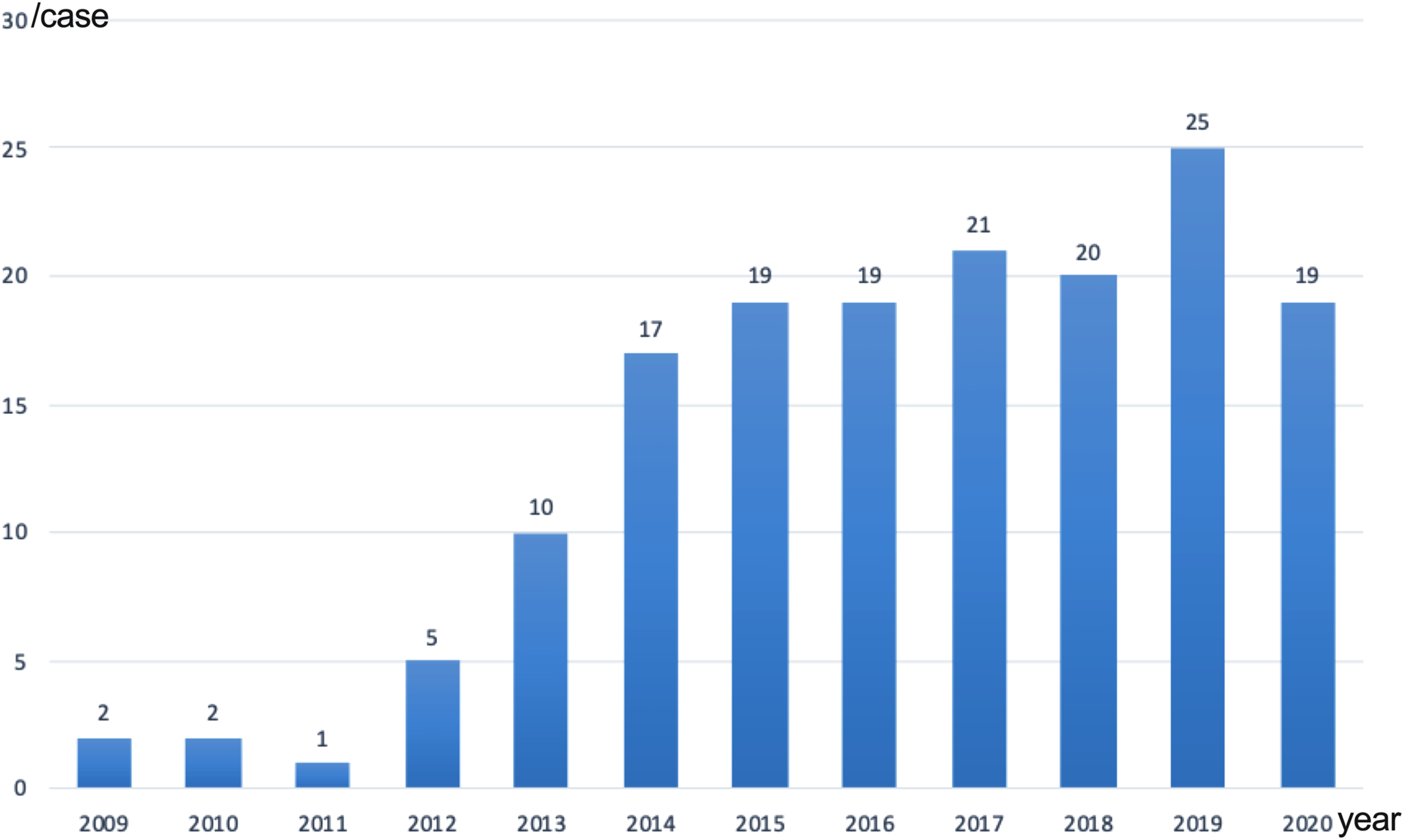
Patient number in each year.
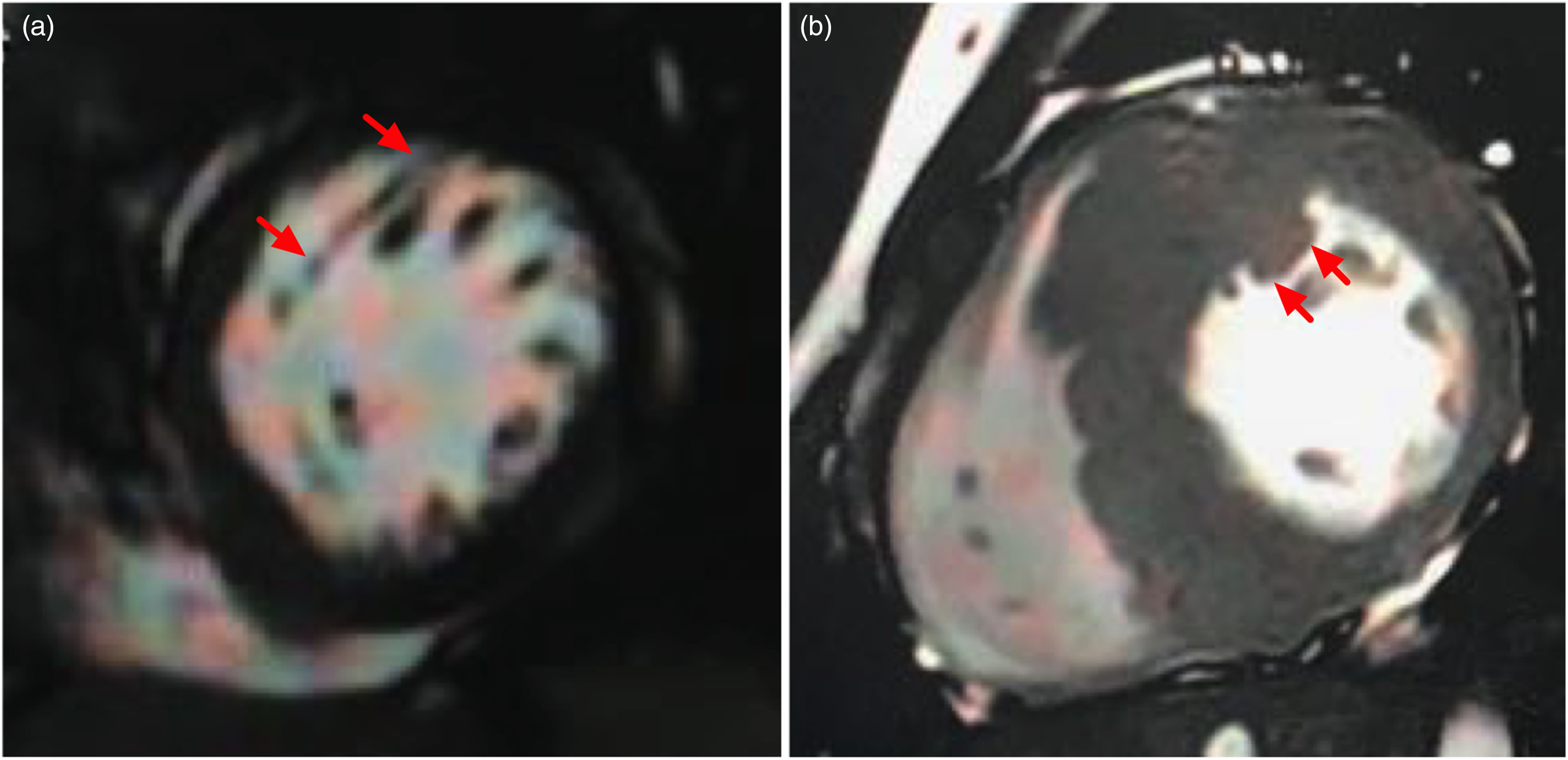
Typical pictures of abnormal left ventricular muscle bundles on cardiac magnetic resonance imaging below. (a) An abnormal left ventricular muscle bundle originates from the root of anterior papillary muscle and extends to anterior ventricular septum in a 16-year-old boy. (b) A thick abnormal left ventricular muscle bundle is attached to anterior ventricular septum closely in a 17-year-old boy (indicated by red arrows).

Representative specimen removed from two child patients with hypertrophic obstructive cardiomyopathy.
Sudden cardiac death and risk stratification
SCD is the most common mode of death outside of infancy in children with HCM, whose annual rates are higher compared with those in adult HCM. 32 Implantable cardioverter-defibrillator (ICD) remains the most effective and reliable treatment to prevent SCD. 33 Nonetheless, device-related complications cannot be neglected in this young patient cohort. Therefore, ICD decisions in children are inevitably involved in the balance of the preservation of life against possible device complications. However, it remains challenging to distinguish patients who are most benefit from an ICD implantation. Recently, the International Pediatric Hypertrophic Cardiomyopathy Consortium has developed an SCD risk prediction model for paediatric HCM using a large, international cohort that provides individualized SCD risk estimates at 5 years. 32 The C index and calibration slope was 0.69 (95% CI, 0.66–0.72) and 0.98 (95% CI, 0.59–1.38), respectively. Thus, the new paediatric SCD risk prediction model shows good discrimination and calibration. This is also the first validated SCD risk score model in childhood HCM. Like the ESC HCM-Risk SCD, this new SCD risk prediction model specified for HCM in children can quantify the individual SCD risk, which could be helpful for clinicians to make an ICD implantation decision in sharing with their parents or guardians.12,34 As the author has pointed out, further refinement of this model may be reasonable by integrating novel imaging markers, such as late gadolinium enhancement in cardiac magnetic resonance.
In summary, HCM in children is rare and a disorder of diverse causes, whose prognosis depends largely on age of presentation and underlying etiology, with patients diagnosed in the first year of life or with inborn errors of metabolism presenting the worst prognosis. In contemporary era, surgical septal myectomy can achieve favourable outcomes for carefully selected paediatric patient in experienced tertiary referral centre. Development of SCD risk prediction model for HCM in children is ongoing, leaving a room for further refinement by adding novel prognostic markers.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Ethical approval
Not applicable.
Informed consent
Not applicable.