
Abstract
Background:
Benzodiazepines bind to γ-aminobutyric acid type A (GABAA) receptor subtypes identified by different α subunits (i.e., α1GABAA, α2GABAA, α3GABAA, and α5GABAA). Sedative-motor effects of benzodiazepines are thought to involve α1GABAA and α3GABAA subtypes.
Aims:
We evaluated observable measures of sedative-motor effects and species-typical behaviors in monkeys following acute administration of novel GABAkines (positive allosteric modulators of GABAA receptors), with varying degrees of selective efficacy at different GABAA receptor subtypes. We predicted that the induction of sedative-motor effects would depend on the degree of α1GABAA and α3GABAA efficacy.
Methods:
Adult female rhesus monkeys (N = 4) were implanted with chronic indwelling i.v. catheters. Quantitative behavioral observation was conducted by trained observers following administration of multiple doses of the conventional benzodiazepine alprazolam and the GABAkines MP-III-80 (preferential efficacy at α2/α3/α5GABAA subtypes), KRM-II-81, MP-III-24 (both with preferential efficacy for α2/α3GABAA subtypes), and MP-III-22 (preferential potency and efficacy for α5GABAA subtypes).
Results:
As with alprazolam, all GABAkines induced significant levels of mild sedation (“rest/sleep posture”). Deep sedation was observed with alprazolam, MP-III-80, and MP-III-22; motoric effects (observable ataxia) were obtained with alprazolam, KRM-II-81, and MP-III-22 only. Surprisingly, the order of potency for rest/sleep posture was significantly associated only with potency at α5GABAA subtypes.
Conclusions:
GABAkines with preferential efficacy at α2/α3GABAA and/or α5GABAA subtypes engendered sedative-motor effects in monkeys, although only compounds with α5GABAA activity engendered deep sedation. Moreover, the significant relationship between potency obtained with in vitro electrophysiology data and the rest/sleep posture measure suggests a role for the α5GABAA subtype in this milder form of sedation.
Keywords
Introduction
Benzodiazepines produce their effects by binding to the γ-aminobutyric acid (GABA) type A (GABAA) receptor, where they increase chloride conductance via positive allosteric modulation (for reviews, see Ghit et al., 2021; Knoflach and Bertrand, 2021). GABAA receptors are heteromeric assemblies of five subunits, with the majority consisting of two α, two β, and one γ subunit. Each subunit family is made up of multiple isoforms (α1-6, β1-3, and γ1-3) with benzodiazepines interacting only with GABAA receptors containing α1, α2, α3, and α5 subunits (α1GABAA, α2GABAA, α3GABAA, and α5GABAA, respectively). Moreover, molecular/genetic and pharmacological studies consistently have shown that the different behavioral effects associated with benzodiazepines (e.g., anxiolysis and sedation) can be linked with distinct GABAA receptor subtypes (for reviews, see Cerne et al., 2022; Engin et al., 2018).
We have developed a behavioral scoring system in rhesus monkeys that incorporates measures of species-typical behaviors as well as levels of sedation derived from anesthesiology standards (e.g., Duke et al., 2018). This behavioral scoring system has been used to address research questions ranging from determining GABAA subtype involvement in sedation (Duke et al., 2018; Meng et al., 2020), to ethanol’s effects on behavior (Berro et al., 2019; Rüedi-Bettschen et al., 2013), to the assessment of the behavioral effects of mu and kappa opioid receptor ligands (Huskinson et al., 2020, 2022). Our research on sedative-motor effects of benzodiazepines, based on experiments with selective and nonselective benzodiazepine ligands, established the working hypothesis that deep sedation (the most robust level of sedation identified) and observable ataxia (i.e., visible signs of motor incoordination) involve primarily the α1GABAA subtype (Duke et al., 2018). In contrast, a milder form of sedation termed “rest/sleep posture,” in which the monkey assumes a posture consistent with sleeping (including closed eyes) but is readily rousable was proposed to be mediated by α3GABAA receptor stimulation (Meng et al., 2020).
Our conclusions about GABAA subtypes and sedative-motor effects were based, with two exceptions, on assessments of ligands that were partial positive modulators of α2GABAA, α3GABAA, and α5GABAA receptors. These included ligands such as TPA023B and MRK-696 (Maramai et al., 2020), which have the interpretational advantage of being neutral or “silent” at α1GABAA receptors. However, it cannot be ruled out that these ligands possess their unique behavioral profiles by virtue of overall reduced efficacy at all subtypes. Our previous studies also included the GABAkines (compounds that are positive allosteric modulators of GABAA receptors) HZ-166 and YT-III-31, which have efficacy profiles that differ from the TPA023B and MRK-696 compounds. In this regard, HZ-166 has relatively high efficacy in vitro at α2GABAA and α3GABAA receptors, yet is a partial modulator at α1GABAA and α5GABAA subtypes (Fischer et al., 2010; Poe et al., 2016). Similar to the selective partial modulators, HZ-166 induced rest/sleep posture but no deep sedation or observable ataxia (Duke et al., 2018). A possible complication with HZ-166 is its poor bioavailability and very low potency (Poe et al., 2016). Because rest/sleep posture tends to occur at the lower dose ranges with the benzodiazepine-type ligands tested to date, a potential explanation for HZ-166’s lack of deep sedation or observable ataxia is that it simply cannot be administered at a high enough dose to engender α1GABAA subtype-mediated effects (Duke et al., 2018). YT-III-31, on the other hand, is a partial-to-full modulator in vitro at α1GABAA, α2GABAA, and α5GABAA receptors, but with efficacy greater than diazepam at α3GABAA receptors (Namjoshi et al., 2013; Meng et al., 2020). In our observation procedure, YT-III-31 induced rest/sleep posture, deep sedation, and observable ataxia. While a seemingly straightforward interpretation of this sedative-motor profile is that the effects are mediated by α3GABAA receptors, we cannot rule out if this compound’s efficacy at α1GABAA receptors was sufficient to induce deep sedation and observable ataxia (rest/sleep posture likely does not involve α1GABAA receptors, since this effect is not sensitive to blockade with an α1GABAA antagonist, Duke et al., 2018). In addition, we cannot rule out a role for partial modulation at α2GABAA and/or α5GABAA subtypes.
To gain a better understanding of the role of differential efficacy at GABAA subtypes in the sedative-motor effects of benzodiazepines, we evaluated four GABAkines with varying degrees of in vitro efficacy (i.e., modulation of chloride currents in cloned receptor systems) but with the common characteristic of having the lowest efficacy at α1GABAA receptors relative to α2GABAA and α3GABAA subtypes (Table 1). Note that “selectivity” or “preferential” in this context does not refer to receptor binding affinity or potency to alter GABA-mediated currents; the compounds are referred to as “preferring” due to having different degrees of efficacy at specific GABAA receptor subtypes. These compounds include KRM-II-81, currently in preclinical development as an anticonvulsant (Cerne et al., 2022), as well as its analogs MP-III-80 (Witkin et al., 2018) and MP-III-24 (Fischer et al., 2017). A key behavioral characteristic of these compounds is a lack of sedative-motor effects in rodent tests, such as the rotarod procedure (Cerne et al., 2022). In addition, we chose a novel imidazodiazepine, MP-III-22, as a ligand with low efficacy at α1GABAA receptors relative to the other subtypes, but with exceptionally high efficacy and potency for the α5GABAA receptors (e.g., Stamenić et al., 2016). This ligand allowed us to explore the possible role of α5GABAA receptors in the behavioral phenomena characteristic of benzodiazepine ligands.
Efficacy and potency of GABAkine compounds in comparison with diazepam, derived from previously published sources. 1 .
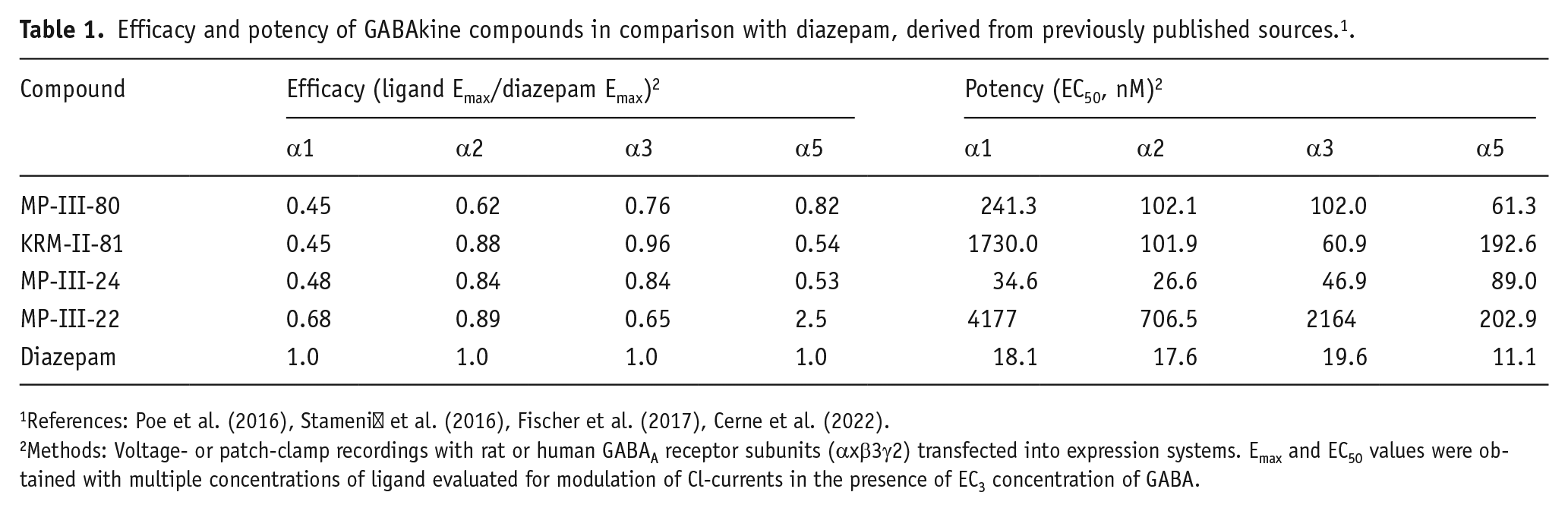
Methods: Voltage- or patch-clamp recordings with rat or human GABAA receptor subunits (αxβ3γ2) transfected into expression systems. Emax and EC50 values were obtained with multiple concentrations of ligand evaluated for modulation of Cl-currents in the presence of EC3 concentration of GABA.
Methods
Animals
Subjects were Indian-origin, adult female rhesus macaques (Macaca mulatta), 14–20 years old, and born at the New England Primate Research Center, Southborough, MA, USA. The monkeys had received test compounds prior to this study, all of which were benzodiazepine-type ligands (Meng et al., 2020; unpublished), and were at least 1-month test free before the study. The monkeys were individually housed and maintained on a 12 h light/12 h dark cycle (lights on at 0600 h), in a vivarium with a temperature of 21°C ± 2°C. The monkeys were maintained on a nutritionally balanced commercial diet (Teklad) supplemented with fruits, vegetables, and forages (dried fruit and seed mix) and had 24 h access to water. All monkeys were prepared with a chronic indwelling venous catheter according to procedures described previously (Platt et al., 2011). The catheter was protected by a stainless-steel tether and nylon jacket (Lomir Biomedical Inc., Malone, NY, USA). All procedures and animal husbandry were conducted according to the Guide for the Care and Use of Laboratory Animals (11th edition), with review and approval by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center.
Drugs/compounds and electrophysiology data sources
Alprazolam was purchased from Tocris Bioscience (Ellisville, MO, USA). KRM-II-81, 5-(8-ethynyl-6-(pyridine-2-yl)-4H-benzo[ƒ]imidazo[1,5-a][1,4]diazepin-3-yl)oxazole; MP-III-80, 3-ethyl-5-(8-ethynyl-6-(pyridine-2-yl)-4H-benzo[ƒ]imidazo[1,5-a][1,4]diazepin-3-yl)-1,2,4-oxadiazole; MP-III-24, methyl 8-ethynyl-6-(pyridine-2-yl)-4H-benzo[ƒ]imidazo[1,5-a][1,4]diazepine-3-carboxylate; and MP-III-22, (R)-8-Ethynyl-6-(2-flurophenyl)-N,4-dimethyl-4H-benzo[ƒ],imidazo[1,5-a][1,4]diazepine-3-carboxamide were synthesized at the Department of Chemistry and Biochemistry, University of Wisconsin-Milwaukee, using previously published methods and with confirmation of purity (Namjoshi et al., 2013; Poe et al., 2016; Stamenić et al., 2016).
Electrophysiological properties of KRM-II-81, MP-III-80, MP-III-24, and MP-III-22 were obtained from published sources (Cerne et al., 2022; Fischer et al., 2017; Poe et al., 2016; Stamenić et al., 2016) and consisted of two-electrode voltage-clamp recording for rat (Xenopus laevis oocyte expression system) or IW-Barracuda automated patch-clamp in human GABAA receptor subunits (HEK-293 cell expression system), normalized to diazepam in each assay. Efficacy values are expressed as the maximum potentiation of GABA-mediated currents as the proportion of the diazepam response for the corresponding subtype.
Although diazepam data was available for the electrophysiology results, we chose alprazolam as a comparative standard for the observation studies. A secondary goal of this report was to provide an internal validity evaluation of our methods since the original data and methods were conducted at a different facility (New England Primate Research Center, NEPRC). We have considerable experience with alprazolam with these methods (Duke et al., 2018, 2020, 2021, unpublished) but have only evaluated diazepam once (Duke et al., 2018). This latter study included alprazolam, and two observations are notable: (1) alprazolam and diazepam engendered a virtually identical profile of behavioral effects and (2) the observable behavioral effects of alprazolam were nearly identical across the two facilities, two different groups of laboratory personnel, and two different groups of monkeys. Based on the above considerations (in particular the replicability of our behavioral measures with alprazolam), we used the Duke et al. (2018) observation data with diazepam to make comparisons between behavioral and electrophysiological results.
Behavioral observation procedure
Behavioral observation sessions were conducted once or twice per week, with at least 2 days between sessions. The behavior of each monkey was scored using a focal animal approach as described by Platt et al. (2002) and modified for rhesus monkeys (Berro et al. 2019; Duke et al., 2018; Huskinson et al., 2020; Rüedi-Bettschen et al., 2013; Sawyer et al., 2014). Parameters and methodological details are provided in Duke et al. (2018; see also Supplemental Materials in Duke et al., 2018).
During a scoring session, a trained observer blind to the drug treatments observed a specific monkey for 5 min and recorded each instance that a particular behavior occurred during 15-s intervals. Scores for each behavior were calculated as the number of 15-s bins in which the behavior occurred (a maximum score would be 20). For sedation measures, structured exposure to stimuli was included in the observation sessions (Duke et al., 2018; for a flowchart-based description, see Figure 1, Huskinson et al., 2020). When a monkey was observed to have closed eyes, an assessment of the monkey’s responsiveness to stimuli was initiated. Specifically, the observer presented three stimuli: (1) walked at a normal pace toward the cage, (2) spoke the animal’s name, and (3) tapped the monkey’s cage with a pen twice. If the monkey responded immediately, that is, opened its eyes and oriented to the observer, rest/sleep posture was scored. If the monkey attended more slowly, that is, opened eyes > 3 s following stimuli), the observer scored moderate sedation. If the monkey did not open its eyes across/throughout the 15 s interval after all three stimuli, the observer scored deep sedation. When scoring moderate or deep sedation, the monkey might be observed to be assuming an atypical posture (e.g., unable to keep an upright posture) that differed from the characteristic rest/sleep posture. The result of this assessment was recorded for the remainder of 15-s intervals in the specific minute of the observation period. If eyes remained closed at the start of the next minute(s), stimuli were again presented in the first 15-s interval of every subsequent minute, the sedation result recorded and carried through the remaining 15-s intervals of the specific minute. If the monkey opened its eyes, sedation measures were no longer recorded. Only eyes closing again initiated the assessment. Note that the maximum score for sedation during a 5-min period was 20 (four possible scores for 4 × 15 s intervals per min, 5 min total; i.e., 4 possible scores × 5 min = 20). The order in which monkeys were observed and the observer performing the scoring each day was randomized. Eight observers participated in the scoring throughout the duration of the study; each observer underwent a minimum of 20 h of training and met an inter-observer reliability criterion of ⩾90% agreement with all other observers for all four monkeys.
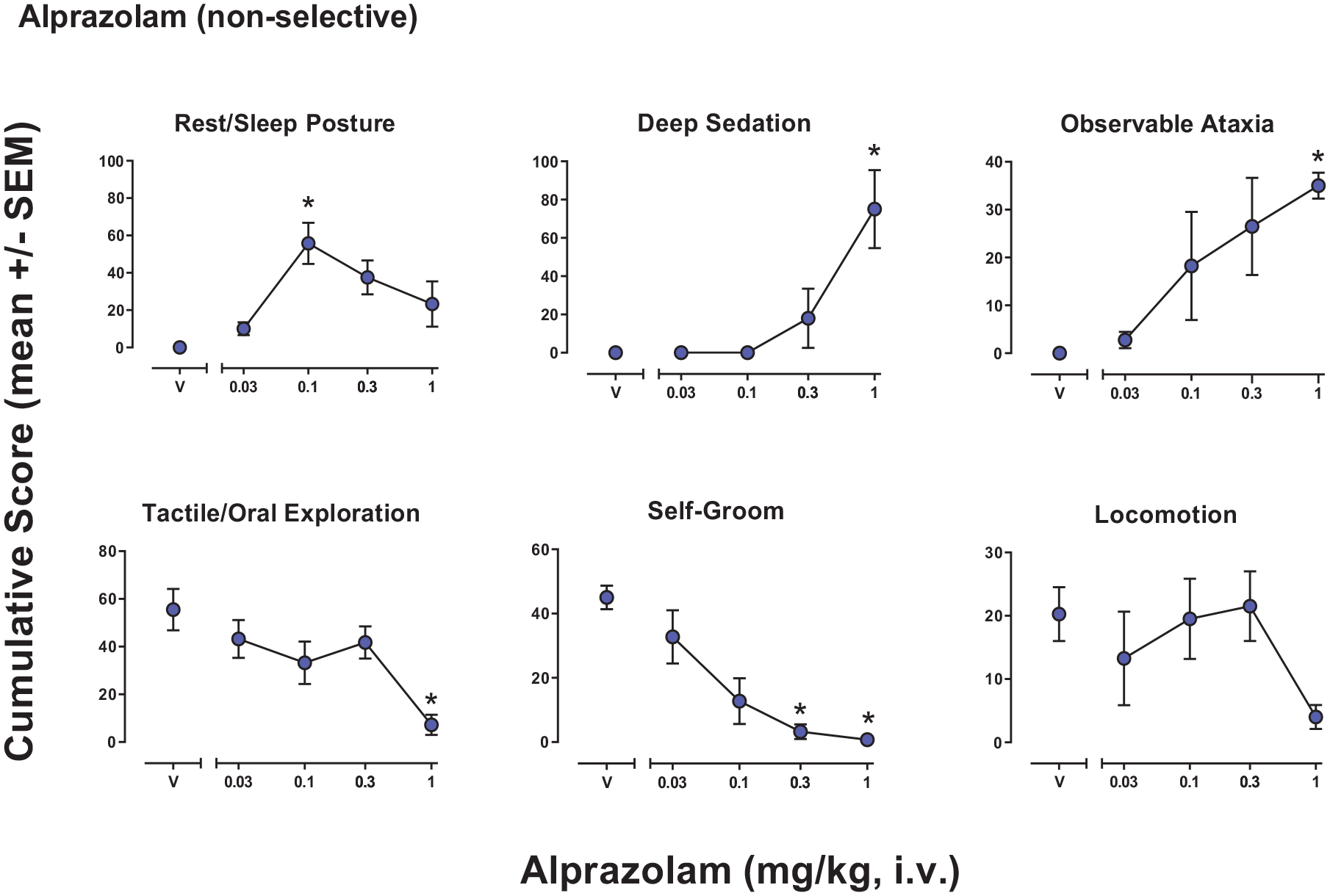
Observable behavioral effects of the benzodiazepine alprazolam in female rhesus monkeys (see Methods, Behavioral observation procedure for definitions). Data are mean modified frequency score (cumulated across multiple time periods, maximum score = 140) ± SEM for n = 4 monkeys.
The 5-min sampling periods were repeated multiple times following i.v. injection of the test ligands or vehicle (min post-injection): 5, 10, 20, 40, 80, 160, and 320. Scores were cumulated across the time periods, resulting in a maximum possible cumulative score of 140 for each behavior. Twenty-four behaviors, including the sedation scores, were included in the scoring protocol, and these behaviors were identified and defined by Duke et al. (2018) and Huskinson et al. (2020). Behaviors in which significant changes occurred in this study, as well as definitions, are as follows: Rest/sleep posture (“idiosyncratic posture adopted by monkeys during rest or sleep, with eyes closed for at least 3 s, easily roused after stimuli presentation”), deep sedation (“atypical loose-limbed posture, eyes closed, does not respond to external stimuli”), observable ataxia (“any slip, trip, fall, loss of balance”), tactile/oral exploration (“any tactile or oral manipulation of the cage or environment”), locomotion (“at least two directed steps in the horizontal and/or vertical plane”), and self-groom (“picking, scraping, spreading, or licking of an animals’ own hair”).
Data analysis
Consistent with our Duke et al. (2018) publication’s supplemental materials, the modified frequency scores in this study were normally distributed (data not shown), so parametric statistics were used. The effects of each ligand were analyzed separately by repeated measures analyses of variance (ANOVA). Treatment effects also were assessed with Bonferroni t-tests in which the effects of different doses were compared with vehicle (because this test controls for family-wise error rates for the pre-selected comparisons, the Bonferroni analyses were conducted irrespective of the ANOVA results). Additional analyses were performed using simple linear regression. For all analyses, the significance levels were set at p ⩽ 0.05. All significant differences had effect sizes (η2G) greater or equal to 0.5, demonstrating that the sample size was sufficient to detect moderate-to-robust effects (consistent with our previously published studies using this methodology, e.g., Duke et al., 2018).
Potency values were calculated for dose-response functions by determining ED50 values, defined as the dose that induced an effect of 50% of the average maximum. The ED50 values were calculated by nonlinear regression (3-parameter logistic) with dose converted to log10 values and cumulative score converted to percent of the maximum. Constrained values included the average score for vehicles and the average maximums. All statistical analyses were performed with GraphPad Prism (Version 9.1.2, build 226, GraphPad Software, Boston, MA, USA).
Results
Summary of electrophysiology re-analysis
Electrophysiology data were available for the four GABAkines studied with purported selectivity for α2GABAA, α3GABAA, and/or α5GABAA receptors (summarized in Cerne et al., 2022; Stamenić et al., 2016) and converted into relative values by dividing Emax and EC50 values by those of diazepam. As shown in Table 1, KRM-II-81 is a partial modulator at α1GABAA and α5GABAA receptors, with Emax values relative to diazepam of 0.45 and 0.54, respectively. KRM-II-81 shows efficacy for α2GABAA and α3GABAA receptors at or near the level of diazepam (0.88 and 0.96, respectively). For the electrophysiology data available for MP-III-80 and MP-III-24, the overall efficacy profiles are similar to KRM-II-81, with some notable differences. For α1GABAA subtypes, MP-III-80 and MP-III-24 are very similar to KRM-II-81, with Emax values approximately half of the Emax for diazepam. MP-III-80 has somewhat lower efficacy at α2GABAA and α3GABAA receptors compared with KRM-II-81, yet nearly diazepam-like levels are apparent with α5GABAA subtypes. Overall, MP-III-24 and KRM-II-81 were highly similar in their efficacy profiles, that is, partial modulator at α1GABAA and α5GABAA but full modulator at α2GABAA and α3GABAA subtypes. In contrast, MP-III-22 has an efficacy profile in vitro that is strikingly different from other GABAkines and benzodiazepines (Table 1). In this regard, efficacies relative to diazepam for the α1GABAA, α2GABAA, and α3GABAA subtypes were above 0.5, ranging from 0.65 to 0.89. However, efficacy relative to diazepam at α5GABAA was 2.5, that is, 250% of diazepam’s efficacy.
Regarding potencies, the data also were derived from the electrophysiology results, with unconverted potency data shown in Table 1. Overall, the most potent GABAkine compound was MP-III-24 (EC50 values of 26.6 to 89 nM across the 4 subtypes). The least potent compound overall was MP-III-22, with its lowest EC50 value being 202.9 at α5GABAA receptors. Differences in potency across subtypes for any given compound tended to be modest, with the largest difference being an approximately 30-fold difference between α3GABAA and α1GABAA subtypes for KRM-II-81.
Behavioral effects of alprazolam (nonselective)
Overall, 6 of the 24 behaviors scored showed significant effects across the test ligands, so all 6 behavioral categories are shown for each ligand. Alprazolam had significant effects for five of the behaviors by either ANOVA or Bonferroni tests, with neither test reaching significance for locomotion (Figure 1). The ANOVA results were: Rest/sleep posture (F(4, 12) = 12.70, p = 0.029), deep sedation (F(4, 12) = 32.58, p = 0.01), observable ataxia (F(4, 12) = 4.56, p = 0.078), tactile/oral exploration (F(4,12) = 5.48, p = 0.036), self-groom (F(4, 12) = 15.41, p = 0.011), locomotion (F(4, 12) = 2.78, p = 0.148). Bonferroni tests showed significant increases or decreases compared with vehicle in cumulative scores that differed across behaviors, with effective doses as follows (mg/kg, i.v.): Rest/sleep posture, 0.1 (increased); deep sedation, 1.0 (decreased); observable ataxia, 1.0 (increased); tactile/oral exploration, 1.0 (decreased); self-groom, 0.3 and 1.0 (decreased).
Behavioral effects of MP-III-80 (α2/3/5GABAA-preferring)
MP-III-80 had significant effects for five of the six behaviors for which at least one dose showed significance versus vehicle, with observable ataxia not reaching significance (Figure 2). The ANOVA results were: Rest/sleep posture (F(5, 15) = 11.79, p = 0.015), deep sedation (F(5, 15) = 14.95, p < 0.001), observable ataxia (F(5, 15) = 2.21, p = 0.107), tactile/oral exploration (F(5, 15) = 2.54, p = 0.074), self-groom (F(5, 15) = 10.91, p < 0.001), locomotion (F(5, 15) = 6.85, p = 0.002). Bonferroni tests showed significant increases or decreases compared to vehicle in cumulative scores that differed across behaviors, with effective doses as follows (mg/kg, i.v.): Rest/sleep posture, 3.0 (increased); deep sedation, 17.0 (increased); tactile/oral exploration, 17.0 (decreased); self-groom, 10.0 and 17.0 (decreased); locomotion, no individual doses (decreased).
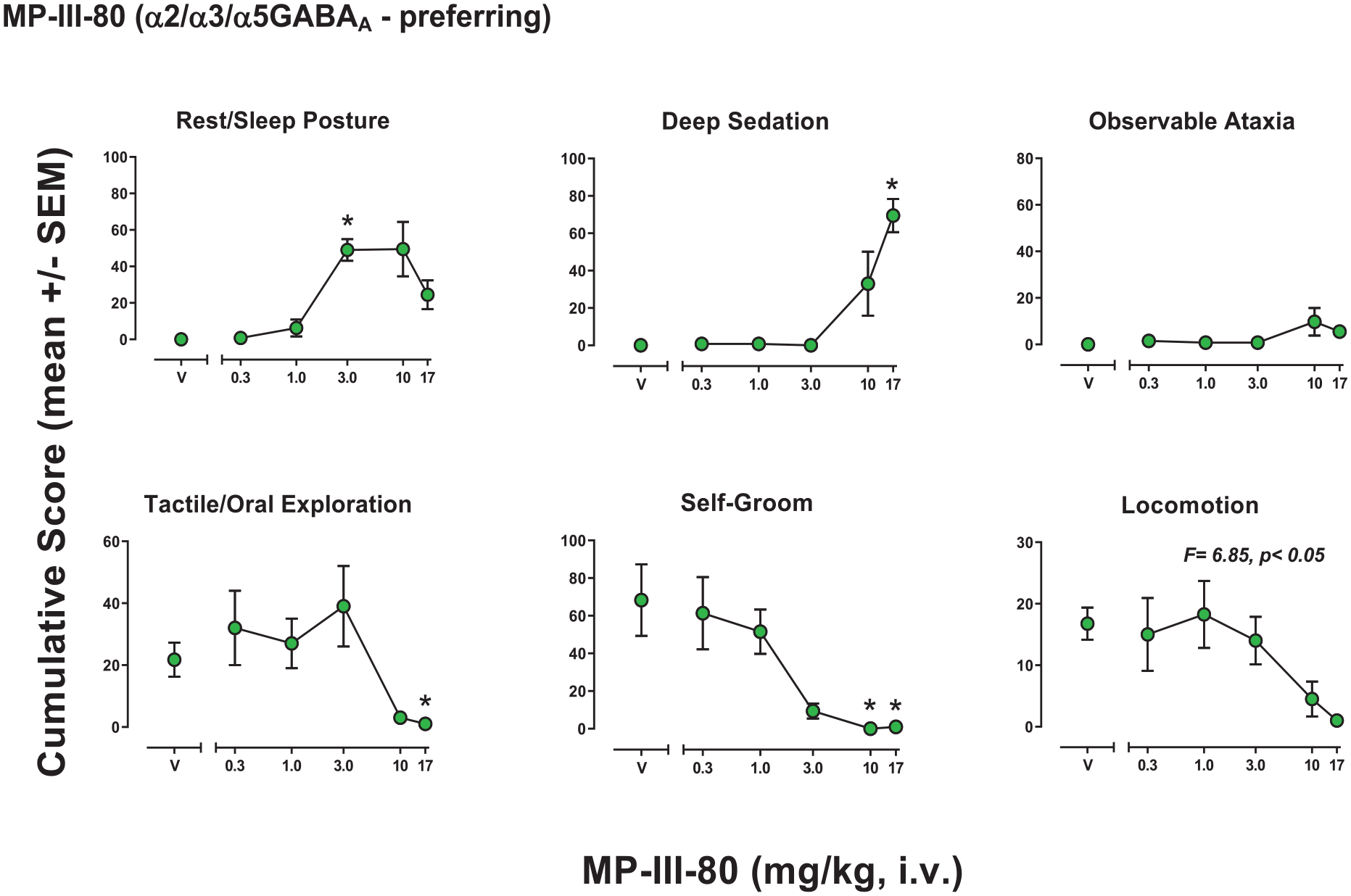
Observable behavioral effects of the GABAkine MP-III-80 (α2/3/5GABAA-preferring) in female rhesus monkeys (see Methods, Behavioral observation procedure for definitions). Data are mean modified frequency score (cumulated across multiple time periods, maximum score = 140) ± SEM for n = 4 monkeys.
Behavioral effects of KRM-II-81 (α2/3GABAA-preferring)
KRM-II-81 had significant effects for five of the six behaviors for which at least one dose showed effects, with no significant effects for deep sedation (Figure 3). The ANOVA results were: Rest/sleep posture (F(5, 18) = 8.64, p = 0.015), deep sedation (F(5, 18) = 0.820, p = 0.489), observable ataxia (F(5, 18) = 4.89, p = 0.010), tactile/oral exploration (F(5, 18) = 3.44, p = 0.019), self-groom (F(5, 18) = 9.15, p < 0.001), locomotion (F(5, 18) = 6.26, p = 0.002). Bonferroni tests showed significant increases or decreases compared with vehicle in cumulative scores that differed across behaviors, with effective doses as follows (mg/kg, i.v.): Rest/sleep posture, no individual doses (increased); observable ataxia, 10.0 (increased); tactile/oral exploration, 17.0 (decreased); self-groom, 10.0 and 17.0 (decreased); locomotion, 1.0 (increased). Of note, the round symbols filled with gray in Figure 2 represent data from a single monkey for which a higher dose could be evaluated due to her relatively small size. Notably, this monkey showed lower levels of rest/sleep posture and observable ataxia, but higher levels of deep sedation, consistent with the dose-response function for deep sedation trending upwards.
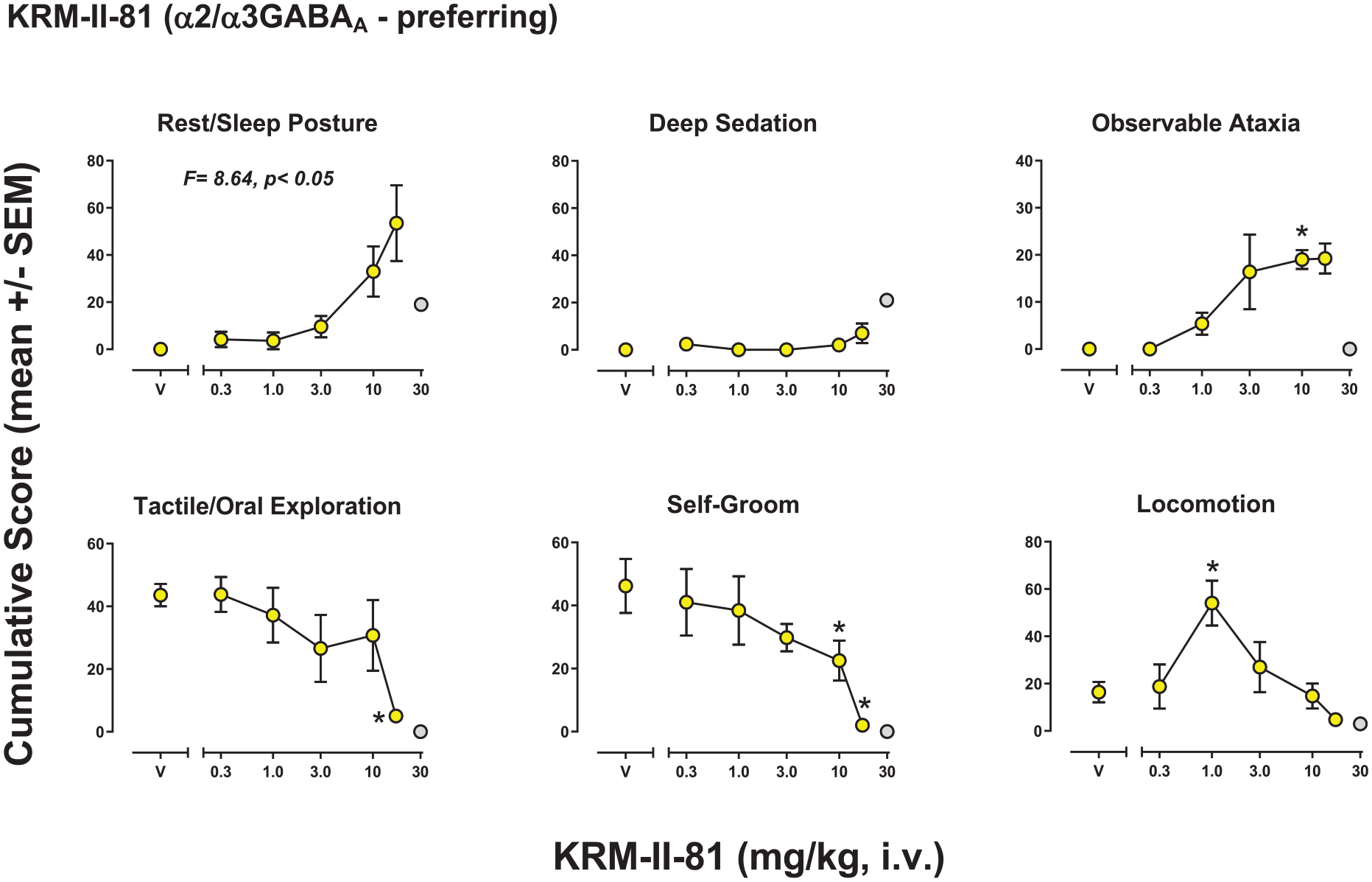
Observable behavioral effects of the GABAkine KRM-II-81 (α2/3GABAA-preferring) in female rhesus monkeys (see Methods, Behavioral observation procedure for definitions). Data are mean modified frequency score (cumulated across multiple time periods, maximum score = 140) ± SEM for n = 4 monkeys, except for the gray-filled circle, which is data from a single monkey.
Behavioral effects of MP-III-24 (α2/3GABAA-preferring)
MP-III-24 had significant effects for only three of the six behaviors for which at least one dose showed effects, making the profile of behavioral effects for this compound strikingly different compared with the others (Figure 4). The ANOVA results were: Rest/sleep posture (F(5, 15) = 33.32, p < 0.001), deep sedation (F(5, 15) = 1.00, p = 0.465), observable ataxia (F(5, 15) = 1.93, p = 0.176), tactile/oral exploration (F(5, 15) = 0.930, p = 0.500), self-groom (F(5, 15) = 10.01, p = 0.001), locomotion (F(5, 15) = 5.20, p = 0.013). Bonferroni tests showed significant increases or decreases compared with vehicle in cumulative scores that differed across behaviors, with effective doses as follows (mg/kg, i.v.): Rest/sleep posture, 1.0 and 10.0 (decreased); self-groom, 17.0 (decreased); locomotion, 10.0 and 17.0 (decreased).
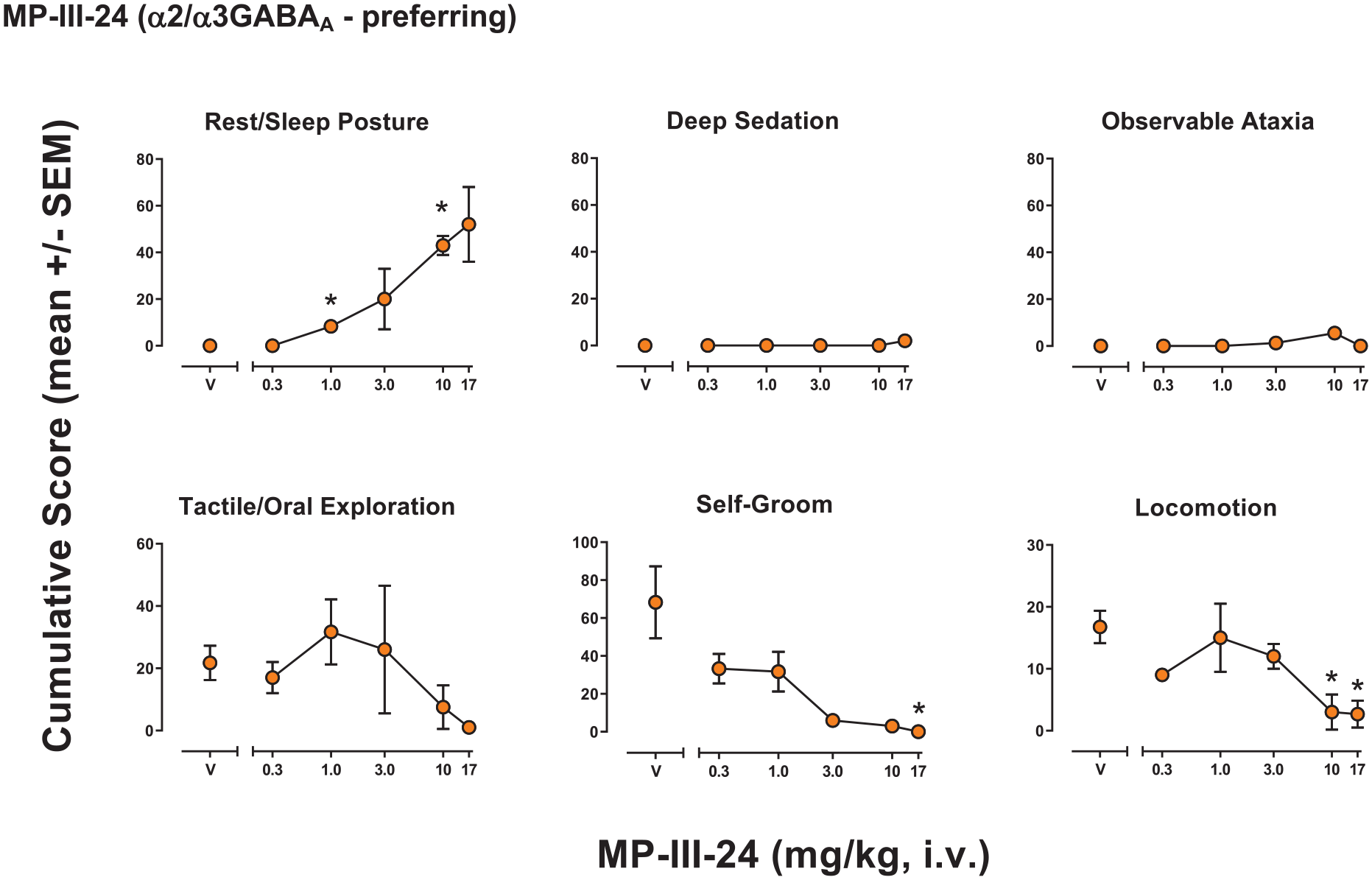
Observable behavioral effects of the GABAkine MP-III-24 (α2/3GABAA-preferring) in female rhesus monkeys (see Methods, Behavioral observation procedure for definitions). Data are mean modified frequency score (cumulated across multiple time periods, maximum score = 140) ± SEM for n = 4 monkeys.
Behavioral effects of MP-III-22 (α5GABAA-preferring)
The α5GABAA selective modulator MP-III-22 had significant effects for five of the six behaviors for which at least one dose showed effects and shared the same significant changes in behaviors seen with alprazolam (Figure 5). The ANOVA results were: Rest/sleep posture (F(5, 15) = 13.90, p < 0.001), deep sedation (F(5, 15) = 50.47, p < 0.001), observable ataxia (F(5, 15) = 16.32, p < 0.001), tactile/oral exploration (F(5, 15) = 4.88, p = 0.008), self-groom (F(5, 15) = 13.21, p < 0.001), locomotion (F(5, 15) = 0.412, p = 0.829). Bonferroni tests showed significant increases or decreases compared to vehicle in cumulative scores that differed across behaviors, with effective doses as follows (mg/kg, i.v.): Rest/sleep posture, 3.0 (increased); deep sedation, 5.6 (increased); observable ataxia, 5.6 (increased); tactile/oral exploration, 5.6 (decreased); self-groom, 5.6 (decreased).
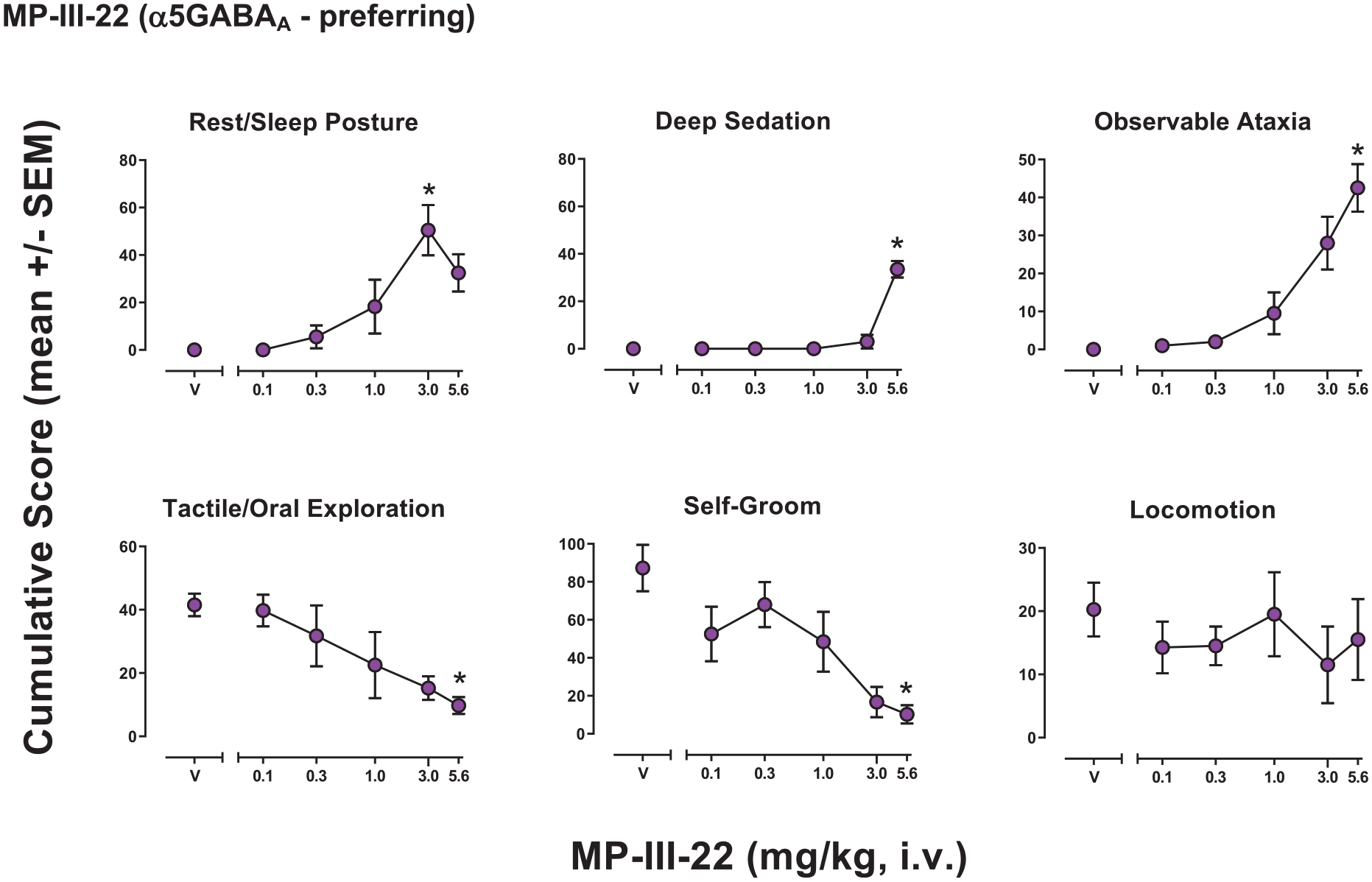
Observable behavioral effects of the GABAkine MP-III-22 (α5GABAA-preferring) in female rhesus monkeys (see Methods, Behavioral observation procedure for definitions). Data are mean modified frequency score (cumulated across multiple time periods, maximum score = 140) ± SEM for n = 4 monkeys.
Behavioral potency comparisons
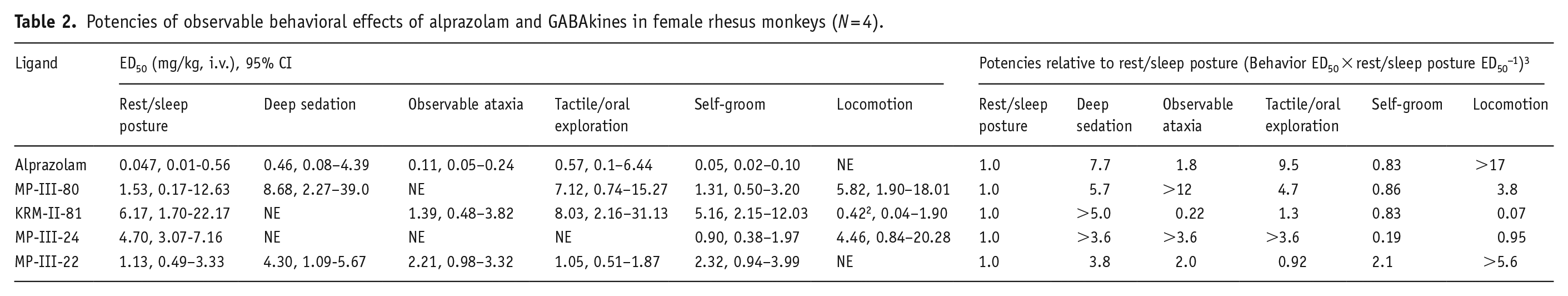
Table 2 shows potencies (ED50 values, obtained from nonlinear regression analysis) for alprazolam and GABAkines across the six behaviors for which at least one significant effect was observed for a ligand. These values also are converted to potencies relative to rest/sleep posture (ED50 for behavior/ED50 for rest/sleep posture), a behavior that was observed for all ligands tested. Data for the conventional benzodiazepine, alprazolam, show a characteristic pattern across the behavioral effects, with the most potent being rest/sleep posture and inhibition of self-groom, whereas the least potent behavior was deep sedation. In general, when deep sedation was observed, it was the least potent behavioral effect of all ligands tested. Interestingly, inhibition of self-groom was equipotent or more potent compared with rest/sleep posture except for MP-III-22, in which inhibition of self-groom required ~2-fold higher doses than rest/sleep posture.
Potencies of observable behavioral effects of alprazolam and GABAkines in female rhesus monkeys (N = 4).
Rank order of potency for behavior versus electrophysiology
To make comparisons between behavioral and electrophysiological results, we used previously published potency data for diazepam based on our observation techniques (Duke et al., 2018) and potency to potentiate GABA-mediated Cl− currents in cloned receptor subtypes (Table 1). All potency measures were normalized to diazepam by dividing the test ligand potency by the diazepam potency (Table 3). Note that inhibition of self-groom was not included in this analysis due to there being no significant decrease in this behavior by diazepam in our prior study (Duke et al., 2018).
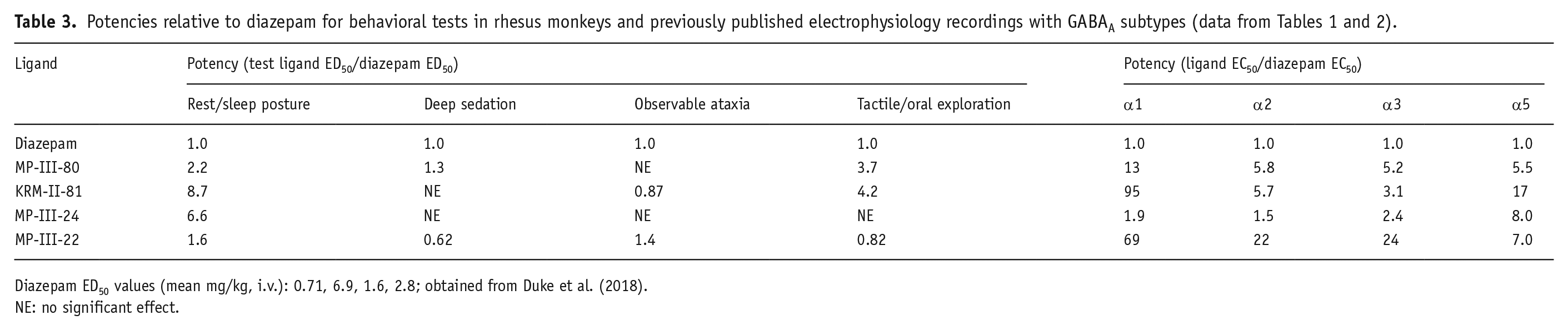
Diazepam ED50 values (mean mg/kg, i.v.): 0.71, 6.9, 1.6, 2.8; obtained from Duke et al. (2018).
NE: no significant effect.
We predicted that the rank order of potency for the GABAkines for inducing rest/sleep posture would show an association with the rank orders of potency at α3GABAA subtypes. To test this prediction, we regressed the potency ratio data (including diazepam as potency ratio = 1.0) using simple linear regression analysis. As shown in Figure 6, associations (R2) for potencies were randomly distributed for α1GABAA, α2GABAA, and α3GABAA. Contrary to our prediction, the R2 value for α5GABAA subtypes was robust (0.82). Linear regression analyses showed that the slope of the fitted line was significantly different from zero for α5GABAA receptors (F(1, 3) = 13.56, p = 0.034), but not α1GABAA (F(1, 3) = 0.69, p = 0.47), α2GABAA (F(1, 3) = 0.32, p = 0.61), or α3GABAA (F(1, 3) = 0.51, p = 0.53) receptors. The regression equation for the α5GABAA subtype was y = 1.55x − 1.06 (y = mx + b; where y = potency ratio for electrophysiology; m = slope, x = potency ratio for rest/sleep posture; b = value of y at origin).
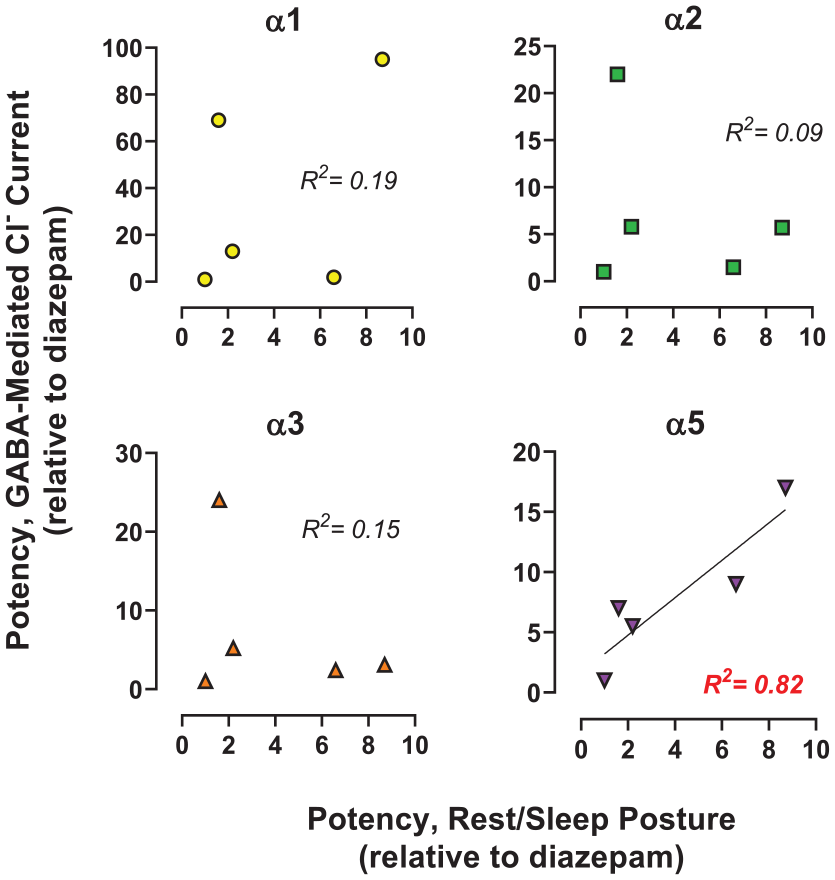
Regression analysis of the relationship of relative potency (vs diazepam) to engender rest/sleep posture and relative potency to potentiate GABA-mediated Cl− currents in cloned rat GABAA receptor subtypes (αxβ3γ2). Each data point is an individual subject. See Table 3 for raw data. R2 values are goodness-of-fit for linear regression analysis, with the values in red font statistically significant versus zero.
Discussion
Our observable behavior scoring system described here and previously has provided an in-depth analysis of the behavioral effects of pharmacological classes of ligands ranging from GABAA modulators to ethanol and opioid receptor ligands (Berro et al., 2019; Duke et al., 2018; Huskinson et al., 2020, 2022; Rüedi-Bettschen et al., 2013). This study extends our previous paper that evaluated novel selective GABAA modulators and revealed evidence for differential roles of the α1GABAA and α3GABAA subtypes in the sedative-motor effects of benzodiazepines (Duke et al., 2018). As in the Duke et al. (2018) report, the present study included the conventional benzodiazepine, alprazolam, as a comparator and demonstrated that this nonselective benzodiazepine-induced rest/sleep posture (a measure of sedation in which the monkey is postured in a manner consistent with sleep, for example, eyes closed, but is immediately roused after presenting stimuli) and, at higher doses, deep sedation (more robust sedation in which the monkey assumes an atypical posture and does not respond to stimuli). Alprazolam also engendered observable ataxia and suppression of the species-typical behaviors of tactile/oral exploration and self-groom. These findings directly replicate (including effective dose ranges) our previous findings reported by Duke et al. (2018), with some exceptions. In this regard, significant moderate sedation was observed by Duke et al. (2018) and not in the present study, whereas decreases in self-groom were not apparent in Duke et al. (2018) yet were significantly attenuated in the present study. These differences reflect increased variability in the moderate sedation measure in this particular set of monkeys, along with relatively higher baseline levels of self-groom in the present monkey cohort compared to the previous animals. The key sedative-motor effects, however, did replicate remarkably well, including relatively similar potency values and order of potencies. For example, the ED50 for rest/sleep posture in Duke et al. (2018) was 0.028 mg/kg, compared with 0.047 mg/kg in the present study, whereas the ED50 for deep sedation in Duke et al. (2018) was 0.59 versus 0.46 mg/kg in the present study. Our primary measure of motor incoordination, observable ataxia, had an ED50 of 0.08 mg/kg in Duke et al. (2018) versus 0.11 mg/kg in the present report. Though not identical, these values were within a 2-fold range across studies with different experimenters, monkeys, and facilities (University of Mississippi Medical Center, present study, vs New England Primate Research Center, Duke et al., 2018), which provides internal validity for our observation techniques.
Our previous report by Duke et al. (2018) evaluated a GABAkine, HZ-166, with a profile of efficacies at the receptor subtypes similar to that of KRM-II-81 and MP-III-24, that is, full modulator at α2/α3GABAA receptors but partial modulator at α1/α5GABAA receptors. MP-III-24 resembled the earlier-tested ligand HZ-166, being devoid of deep sedation and observable ataxia. However, KRM-II-81 and its analog, MP-III-80, engendered observable ataxia and deep sedation, respectively. The Duke et al. (2018) study demonstrated that deep sedation and observable ataxia could be antagonized by an α1GABAA-selective antagonist. This previous study also included the compounds MRK-696 and TPA023B, which have a different profile in that they are partial modulators at α2/α3/α5GABAA receptors, with TPA023B having no efficacy at α1GABAA receptors (Maramai et al., 2020). These compounds did not induce deep sedation, with MRK-696 engendering observable ataxia that was attributed to partial modulation at α1GABAA receptors. Based on these prior results collectively, we concluded that α1GABAA receptors likely mediate deep sedation and observable ataxia-associated benzodiazepine-like ligands. However, the findings of observable ataxia with KRM-II-81, as well as deep sedation with MP-III-80, cast some doubt on this hypothesis.
A possible alternative explanation for the findings with HZ-166 and MP-III-24 is that the ligands were not potent enough to occupy a sufficient number of α1GABAA receptors to induce sedative-motor effects (see Sieghart and Savić, 2018, for discussion of this hypothesis). Indeed, HZ-166 has relatively low potency both in vivo and in vitro. However, MP-III-24 was the most potent of the compounds in vitro in the present study (note, however, that these results are based on potency in electrophysiology assays rather than binding affinity, a more direct assessment of in vitro potency). Moreover, KRM-II-81 was the least potent at α1GABAA receptors in vitro, yet induced significant observable ataxia and a trend toward deep sedation. These observations controvert the explanation that bioavailability was a major determinant of functional selectivity for these compounds, although it should be noted that neither KRM-II-81 nor MP-III-80 resulted in both deep sedation and observable ataxia, in contrast to the nonselective full modulators tested to date (cf. Duke et al., 2018). Therefore, it may be the case that α1GABAA receptor-mediated effects are expressed inconsistently with compounds acting as partial modulators at the α1GABAA subtype, that is, full modulators may be more likely to engender both observable ataxia and deep sedation. This inconsistency may reflect that a threshold level of intrinsic efficacy needs to be reached to express each behavior, and partial modulators have efficacy close to the threshold.
Of the GABAkines evaluated to date, KRM-II-81 may have the most unique profile. In this regard, KRM-II-81 induced observable ataxia at doses lower than those that engendered rest/sleep posture yet did not result in deep sedation over the dose range tested. Because rest/sleep posture potencies are correlated with anxiolytic-like potencies in rhesus monkeys (Duke et al., 2018), these observations are consistent with results from rodent procedures suggesting KRM-II-81 has anxiolytic-like effects at doses not associated with sedative effects (for review, see Cerne et al., 2022). Moreover, suppression of self-groom tended to be the effect for which other compounds were most potent, whereas KRM-II-81 was considerably less potent with this behavior relative to other behaviors. Finally, KRM-II-81 enhanced, rather than decreased, locomotion. A caveat to this profile is that due to solubility limits, we were unable to increase doses except in one smaller monkey. For this monkey, moderate levels of deep sedation were apparent, along with suppression of locomotion. As mentioned above, KRM-II-81 showed micromolar potency at α1GABAA receptors in electrophysiology studies, consistent with the idea that an α1GABAA partial modulator would need high levels of occupancy to show consistent effects, which in this case may have been limited by solubility constraints.
Another unique ligand evaluated in the present study was MP-III-22, a GABAkine described previously as having preferential efficacy for α5GABAA subtypes (Stamenić et al., 2016). Indeed, MP-III-22 showed significantly higher efficacy at α5GABAA subtypes in vitro than diazepam and was the most potent at potentiating GABA-mediated currents at that receptor. However, MP-III-22 displayed a profile of observable behavioral effects that were more similar to that of alprazolam than with the other GABAkines. This is perhaps not surprising, given the efficacy of this compound at the other GABAA subtypes was relatively close to that of diazepam, making it likely that all receptors would be activated at a high enough dose. In this regard, Stamenić et al. (2016) showed, via a combination of behavioral studies, electrophysiology, and binding site occupancy data, that higher doses of MP-III-22 lost selectivity in rats. Moreover, the dose range for selectivity was relatively narrow (Stamenić et al., 2016), raising the possibility that the behavioral profile of MP-III-22 simply reflected a loss of selectivity in monkeys.
Interestingly, MP-III-22 has micromolar potency at α1GABAA subtypes and is approximately 69-fold lower in potency than diazepam at this site, yet was almost equipotent with diazepam in its ability to engender deep sedation. Although we do not have data on binding site occupancy with these compounds, these discrepancies in potency in vitro versus in vivo are inconsistent with this subtype mediating this behavior. Although based on our previous findings (Duke et al., 2018), deep sedation likely is α1GABAA-mediated, the mechanisms conceivably may be different with the higher-efficacy GABAkines, that is, higher efficacy may reveal roles of other receptor subtypes. In this regard, it is notable that only ligands with relatively high efficacy at α5GABAA receptors (alprazolam, MP-III-80, MP-III-22) engendered deep sedation. Although entirely correlative, the possibility that α5GABAA receptors play a role in deep sedation warrants further investigation.
To date, all GABAkines, including the Merck compounds with very different in vitro efficacy profiles (e.g., TPA023B and MRK-696, Duke et al., 2018) from the compounds tested here, engendered significant levels of rest/sleep posture, characterized as a relatively mild form of sedation. We previously attributed this effect to α3GABAA receptors and not α1GABAA receptors due to (1) rest/sleep posture being common to all compounds with reduced or no efficacy at α1GABAA receptors, (2) rest/sleep posture being insensitive to an α1GABAA-selective antagonist, and (3) rest/sleep posture occurring after administration of an α3GABAA-preferring positive modulator (Duke et al., 2018; Meng et al., 2020). In the present study, we regressed the potencies of the GABAkines to engender rest/sleep posture versus the potencies at the four GABAA subtypes to modulate Cl− currents in the presence of GABA. Counter to our predictions, we found a robust effect with the α5GABAA subtype only. This finding was surprising, given that HZ-166 and MP-III-24 were considered to have preferential efficacy for α2/3GABAA subtypes, with lower efficacy at α5GABAA subtypes. However, it is important to note that both compounds do have partial modulatory effects at α5GABAA subtypes, as is the case for the Merck compounds TPA023B and MRK-696, which also engendered robust rest/sleep posture (Duke et al., 2018). To date, we have not examined observable behavioral effects of compounds lacking efficacy entirely at α5GABAA subtypes. Interestingly, the closest such ligand we’ve tested is α1GABAA-selective drug, zolpidem, which does not bind to α5GABAA subtypes. Consistent with our new hypothesis, zolpidem lacked significant rest/sleep posture (Duke et al., 2018).
In rodent brains, the α5GABAA subtype is highly expressed in the hippocampus and olfactory bulb, with relatively low or no detectable levels in other brain regions and moderate-to-strong expression in the spinal cord (Fritschy and Mohler, 1995; Sperk et al., 1997). In contrast, in the rhesus monkey brain, the α5GABAA subtype is much more ubiquitous, with robust expression in the cortex, basal ganglia, amygdala, and thalamus (Sperk et al., 2020; spinal cord expression in monkey is unknown at present). The function of the α5GABAA subtypes in rodents appears to be complex, ranging from an expected role in cognition (e.g., Collinson et al., 2002) to an unexpected role in anxiolysis (Behlke et al., 2015; Botta et al., 2015). Given the broader distribution of the α5GABAA receptors in primate brains compared to rodent brains, it is perhaps unsurprising that this subtype may be functionally complex in primates as well, potentially playing a key role in sedative effects induced by benzodiazepine-type ligands. However, as described above, MP-III-22 suppressed locomotor activity and rotarod performance in rats but only at higher doses not associated with selective activation of the α5GABAA receptor (Stamenić et al., 2016). On the other hand, Savić and colleagues (Milić et al., 2012; Savić et al., 2008) have demonstrated, via use of other α5GABAA subtype-preferring compounds, evidence in rats that this receptor subtype may play a role in benzodiazepine-associated myorelaxant effects and locomotion suppression. These authors posited a role for spinal α5GABAA subtypes, which may be the case in the present study, although the distribution of α5GABAA subtypes in rhesus monkey spinal cord is not yet known.
As mentioned above, our studies do not directly address pharmacokinetic factors, such as the possibility of active metabolites, resulting in the presence (or absence) of deep sedation and observable ataxia. To help negate the influence of pharmacokinetic factors, all compounds were administered i.v. and the observation procedure uses time sampling. Maximal effects were observed early in the session and maintained across the entire observation period for the four GABAkines (data not shown). Moreover, the differences among compounds primarily were qualitative in nature, i.e., different compounds had distinctive behavioral profiles, rather than quantitative differences, that is, the GABAkines did not show differences primarily in the strength of behavioral effects. The latter pattern of effects might be expected if the compounds differed in factors such as brain penetrability. It also is important to note that none of the GABAkines had unequivocal selective efficacy, that is, all compounds had some modulatory action at all GABAA receptor subtypes, raising the question of to what extent the unique sedative effects represented interactions among the subtypes. Finally, other GABAA binding sites exist that may very well play a role in the findings, for which insufficient data (and selective compounds) exist for systematic study at the present time (e.g., Sieghart et al., 2012).
The translation of preclinical measures of sedation to clinical assessments of sedative effects associated with benzodiazepine ligands has proven to be challenging. For example, the partial GABAA modulator MRK-409 lacked sedative-motor effects in rats and monkeys, yet engendered pronounced sedation in human subjects (Atack et al., 2009). This effect was attributed to MRK-409 having efficacy at the α1GABAA subtype; however, subsequent evaluation of ligands that were antagonists or had very low efficacy at α1GABAA receptors has shown at least modest behavioral effects associated with sedation (e.g., adverse events reported as “dizziness,” “somnolence”; Atack et al., 2010; Nickolls et al., 2018). We have addressed this translational concern by using observational studies in monkeys, and consistent with clinical findings, have also demonstrated modest levels of sedation with ligands lacking α1GABAA efficacy (Duke et al., 2018). Moreover, a compound (MRK-696) with an efficacy profile similar to that of MRK-409 was found to engender observable ataxia and rest/sleep posture in monkeys (Duke et al., 2018), consistent with the observations of sedative-motor effects in human subjects. Therefore, we propose that the lack of detection of sedative effects in preclinical models may have much to do with the methodology used, rather than just species differences in GABAA subtype efficacy/selectivity. Interestingly, the present study raises the possibility that a compound with zero efficacy at the α5GABAA subtype may lack even modest levels of sedation. Nevertheless, a key takeaway from these studies is that novel GABAkines may at least have reduced sedative-motor effects (although not completely absent) in humans, which offers a significant improvement over anxiolytic benzodiazepines commonly prescribed today.
Footnotes
Acknowledgements
We acknowledge the University of Wisconsin-Milwaukee’s Shimadzu Laboratory for Advanced and Applied Analytical Chemistry and support from the Milwaukee Institute of Drug Discovery.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported by USPHS grants DA011792, DA043204, DA049886, DA052801, DA058666, AA029023, AA029306, and GM121334.