
Abstract
Introduction:
We present the management of the second reported case of carcinosarcoma of the kidney parenchyma with malignant degeneration of both epithelial and mesenchymal components.
Case report:
A 36-year-old woman came to our attention for an incidental sonographic finding of a lesion in the lower pole of the left kidney. A contrasted computed tomography scan confirmed the presence of a 50 × 52 mm2 contrasted lesion in the left lower kidney pole. The patient underwent a challenging laparoscopic left partial nephrectomy and para-aortic lymphadenectomy. The histological examination led to the diagnosis of a carcinosarcoma of the kidney parenchyma with malignant degeneration of both epithelial and mesenchymal components. The patient underwent adjuvant chemotherapy with paclitaxel at 175 mg/m2 plus carboplatin area under the curve 6 intravenously for six cycles. At a follow-up of 42 months, the patient is alive and does not show any local recurrences or distant metastases.
Conclusion:
A multi-disciplinary therapeutic approach, combined with an adequate doctor–patient relationship and a close and detailed follow-up, is of fundamental importance in obtaining good outcomes in such rare and challenging cases.
Introduction
Carcinosarcomas are highly aggressive biphasic tumors, made of both a stromal and an epithelial component, that very rarely affect the urinary tract. 1 While most mixed epithelial and stromal tumor (MEST) lesions are benign, cases with malignant transformation may be diagnosed in various organs such as uterus and ovaries. Few cases have been reported in the literature regarding carcinosarcoma arising from the kidney pelvis,2,3 while anecdotal is the emergence of these tumors from the renal parenchyma.
When a physician has to face such unexpected case, he has to deal with the high aggressiveness of the tumor itself, the great heterogeneity of the carcinomatous and sarcomatoid components, and the absence of any clinical trial or guidelines that can help him to lead its management. What is known from the few published cases is that regardless of the type of the primary and adjuvant treatments, the prognosis is poor, with the reported longest survival of about 2 years. 4
We present the case of a patient who has undergone a laparoscopic partial nephrectomy with a histological diagnosis of carcinosarcoma and the following management at a 3.5 years’ follow-up. To the best of our knowledge, this is the second reported case of carcinosarcoma of the kidney parenchyma with malignant degeneration of both epithelial and mesenchymal components.
Case description
A 36-year-old woman with no previous significant medical history came to our attention at the beginning of 2016 for an incidental sonographic finding of a lesion in the lower pole of the left kidney. She did not complain of hematuria, flank pain, or any other symptoms. Blood tests were in the following range: Hb, 9.8 g/L; creatinine, 0.9 mg/dL; estimated glomerular filtration rate, 104 mL/min. A contrasted computed tomography (CT) scan confirmed the presence of a 50 × 52 mm2 contrasted lesion in the left lower kidney pole. The lesion showed the presence of necrosis and adherence both to the calyceal system and to the perirenal fat (Figure 1) (PADUA 11; RENAL 10).
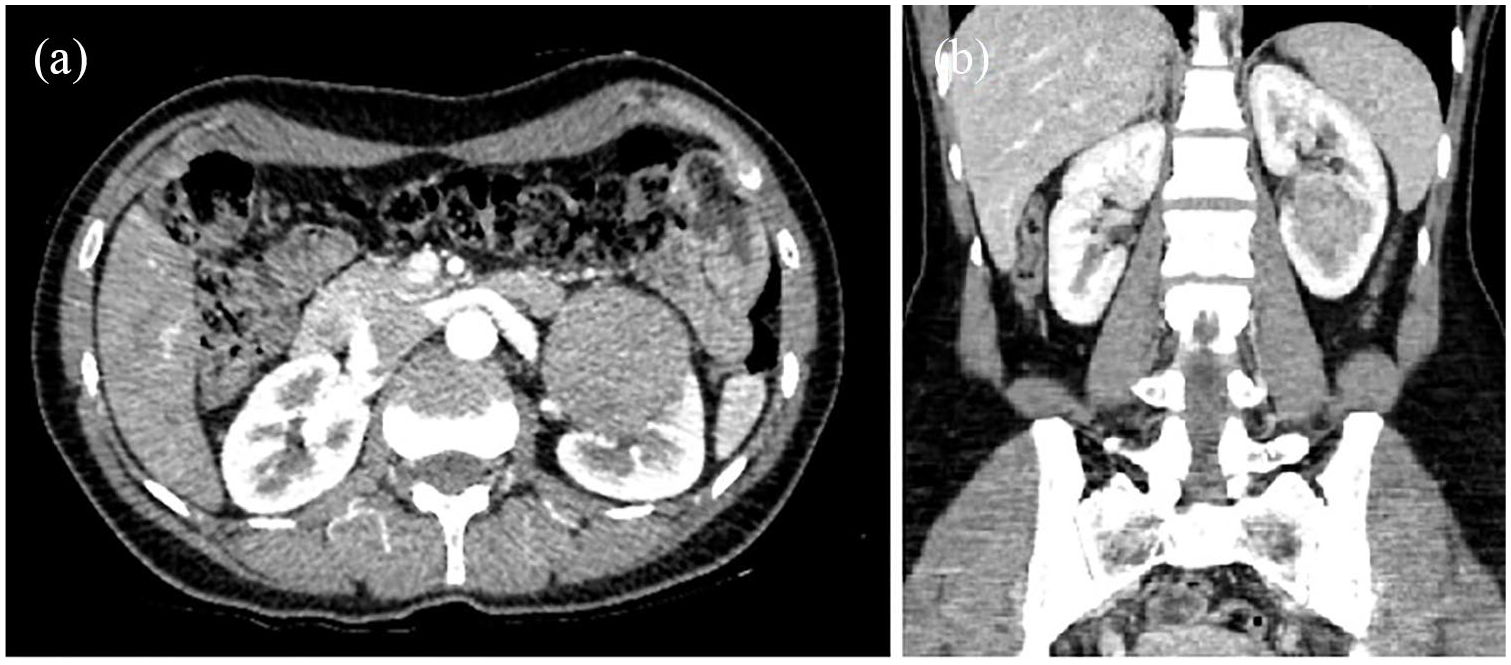
CT scan images showing the presence of a 50 × 52 mm2 lesion in the left lower kidney pole ((a) and (b)).
The patient underwent a challenging laparoscopic left partial nephrectomy and para-aortic lymphadenectomy. The operative time was 210 min with a blood loss of 150 mL. During the postoperative period, the patient was started on antibiotics for postoperative fever and was discharged on postoperative day 6. On histological examination, the aforesaid lesion was well circumscribed with negative surgical margins. The lesion featured both epithelial and stromal components variably intermixed. The epithelial component ranges from tubules to solid nests with low-cuboidal epithelial cells; the stromal component was variably cellular and composed of spindle cells. Both components showed malignant transformation. Carcinomatous area with infiltrative margins, cytological atypia, and occasional mitoses showed unclassifiable, not otherwise specified, features and metastasized to one para-aortic lymph node. Moreover, stromal overgrowth with hypercellularity, atypia, mitoses, and a necro-hemorrhagic area was observed consistent with sarcomatous degeneration (Figures 2 and 3). Proliferative activity was evaluated by Ki-67 monoclonal antibody: 5% and 30% in the epithelial and carcinomatous component, respectively.
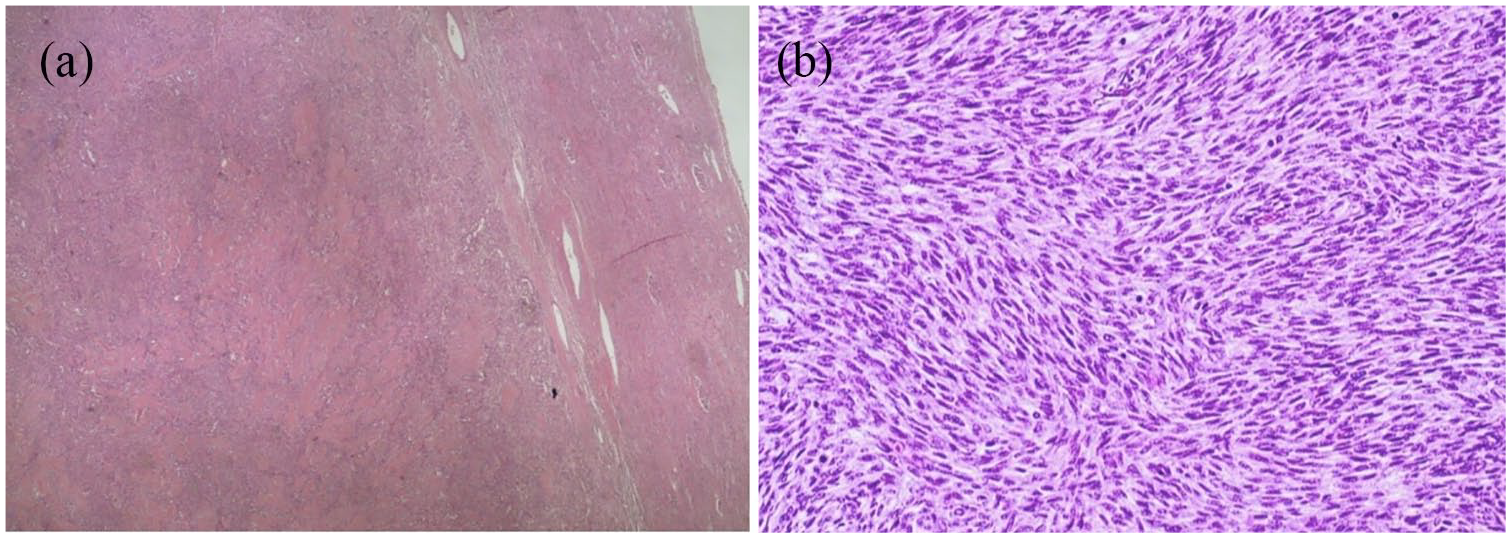
(a) This microphotograph shows the relationship between the neoplasm and the surrounding kidney parenchyma. (b) Sarcomatous degeneration was observed, with fascicular arrangement of spindle cells, without heterologous differentiation. Note the hypercellularity and the plenty of mitosis.
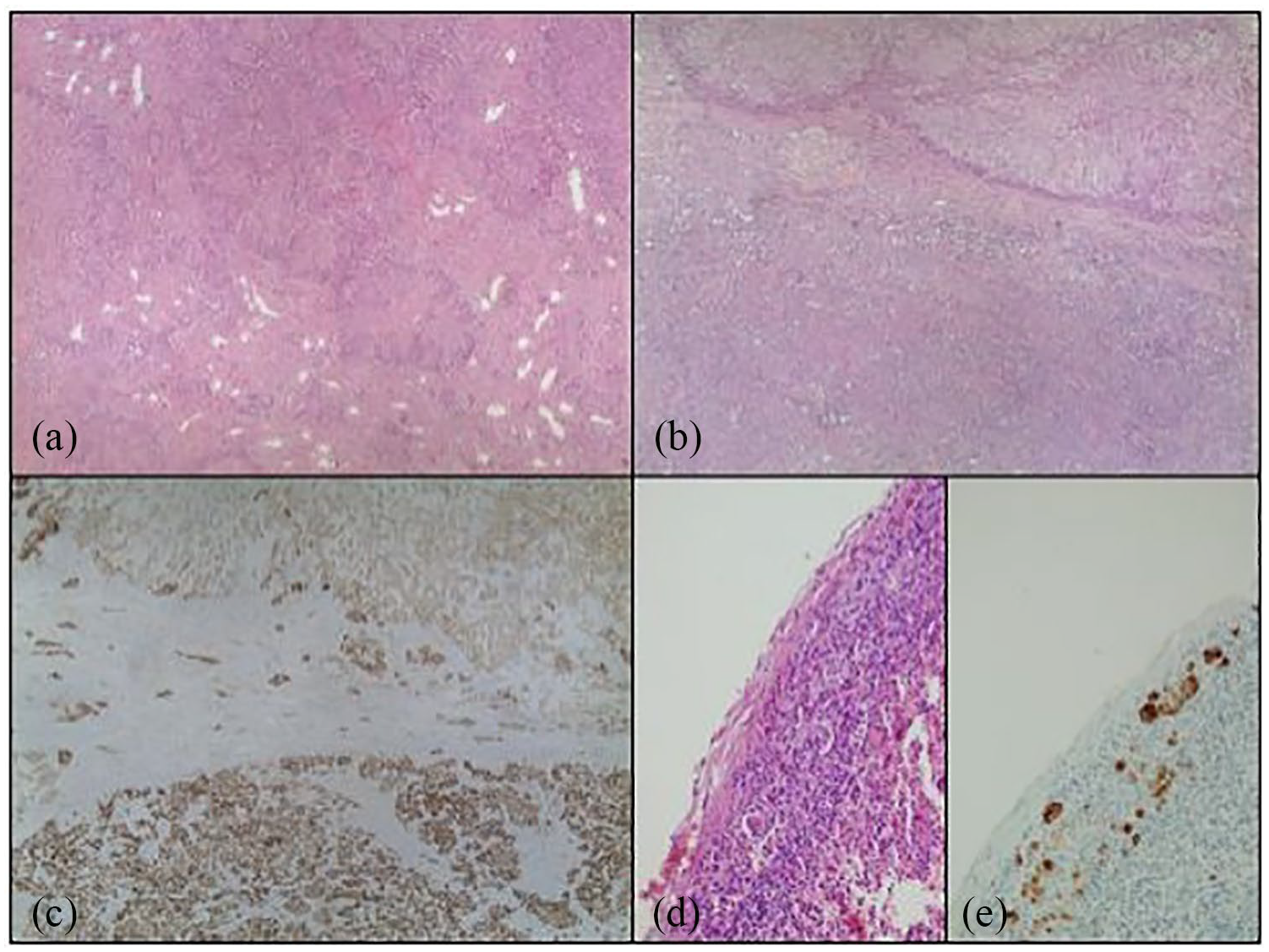
The neoplasm was largely composed of organoid fibroepithelial structures (like those shown in Figure 2(a), hematoxylin and eosin (H&E), 40×). However, the tumor showed malignant degeneration of both the epithelial (low part of Figure 2(b), H&E, 40× and Figure 2(c), CAM5.2, 40×) and stromal components (see Figure 3). The carcinomatous component of the tumor was metastatic to one regional lymph node (Figure 2(d), H&E, 100× and Figure 2(e), CAM5.2, 100×).
After a multi-disciplinary team (MDT) discussion, negative postoperative staging, and careful counseling, the patient, 8 weeks after surgery, underwent adjuvant chemotherapy with paclitaxel at 175 mg/m2 plus carboplatin area under the curve 6 intravenously for six cycles.
At a follow-up of 42 months, the patient is alive and does not show any local recurrences or distant metastases.
Conclusion
Managing a rare disease is always a challenge, especially if there are no treatment guidelines or evidence-based recommendations. The MEST lesions are biphasic neoplasm consisting of mesenchymal and epithelial components. Malignant transformation of the mesenchymal component is seldom encountered, but the occurrence of malignant degeneration of both epithelial and mesenchymal components is even more rare. To the best of our knowledge, this scenario was previously reported only once in English literature, 5 even though also the malignant degeneration of only one of the two components is still a very rare condition.
The surgical approach toward these rare neoplasms should be radical, with the achievement of large negative margins. However, there are no radiological features that allow surgeons to distinguish these tumors preoperatively from a renal cell carcinoma (RCC) and opt for a more aggressive approach.
Due to the complexity of the case and the lack of evidence, an MDT discussion and a careful counseling of the patient become mandatory to design further management and follow-up. Considering the very poor oncological outcomes reported in similar cases,4,5 with the early tendency to develop metastasis, in agreement with the patient we decided to go for an adjuvant chemotherapy.
Because of the rarity of the neoplasm, there is no guideline regarding chemotherapy treatment. We chose the treatment according to few literature data. Unfortunately, the only available experiences concern the carcinosarcoma of ovary and uterus. This chemotherapy regimen is the best tolerated and effective in patients with carcinosarcoma, also in adjuvant setting.6,7
Adjuvant chemotherapy is advisable in patients with advanced stages (III, IV) of carcinosarcoma of the uterus after surgery. 6 In this case, we decided to translate the few data available into our clinical experience. The therapy was well tolerated by the patient without main side effects.
Given the exceptional rarity and aggressiveness of the tumor, we designed a very strict follow-up compared to the standard one of RCC: the first chest-abdomen CT scan was planned at 3 months and then every 6 months for the fists for 3 years. Every 3 months for the first 3 years, we have evaluated the patient with a clinical visit, abdominal sonography, and standard blood examinations. We plan to perform a CT scan annually and a semiannual clinical/laboratory evaluation for the next 2 years.
A multi-disciplinary therapeutic approach, combined with an adequate doctor–patient relationship and a close and detailed follow-up, is of fundamental importance in obtaining good outcomes in such rare and challenging cases.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.