
Abstract
A male infant with trisomy 21 simultaneously showed clinical features of hypomelanosis of Ito and hemimegalencephaly, with related intractable epileptic seizures. The epileptic seizures were refractory to conventional antiepileptic drugs and persisted until the patient underwent functional hemispherotomy. It is well known that patients with hypomelanosis of Ito may also have cortical dysplasia and hemimegalencephaly and that approximately half of these patients have chromosomal abnormalities. However, to our knowledge, there is no previous report of a patient with trisomy 21 associated with hemimegalencephaly. Here, we describe a rare case of coexisting trisomy 21 and hypomelanosis of Ito, associated with hemimegalencephaly.
Keywords
The main complications associated with trisomy 21 include intellectual disability, hypothyroidism, cardiac malformation, duodenal atresia, leukemia, and epilepsy. 1,2 Approximately 7% to 9% of patients with trisomy 21 have epilepsy, while a percentage also suffer from West syndrome. 1 –4 However, the epileptic seizures are associated with a relatively good prognosis, even in patients with West syndrome. 3,4 Furthermore, there has been no report of a patient with trisomy 21 who also had distinct cortical dysplasia or hemimegalencephaly, with related refractory epilepsy. In contrast, it is well known that hypomelanosis of Ito is often associated with cortical dysplasia, hemimegalencephaly, various chromosomal abnormalities, and refractory epileptic seizures. 5,6 We examined a male infant with concurrent clinical features of trisomy 21 and hypomelanosis of Ito, who had refractory epilepsy due to hemimegalencephaly. We herein describe a rare case of coexisting trisomy 21 and hypomelanosis of Ito, and the clinical features.
Case Report
A male infant started showing right-sided facial twitching with secondary right-sided unilateral and generalized tonic-clonic seizures at 5 months of age. When his seizure frequency increased, he was transferred to our hospital. His parents were unrelated, healthy, 26-year-old Japanese. The patient had been born after 39 weeks and 2 days of gestation, and his intrauterine period and birth were uneventful. At birth, his measurements were as follows: height, 46.4 cm; weight, 3080 g; chest circumference, 31.0 cm; and head circumference, 33.0 cm. He showed no asphyxia at birth; however, immediately after birth, stridor, hypotonia, and poor sucking were observed. On the basis of his facial appearance, he was suspected to have trisomy 21, and this was later confirmed by chromosomal G-band staining (47,XY, +21). Neither tube feeding nor artificial respiratory management was required. Smile and visual pursuit were observed at 2 months of age, but these were lost after the onset of seizures. At 5 months of age, the patient still had no head control. It was at this time that he was transferred to our hospital for the management of recurrent secondary generalized tonic-clonic seizures lasting 1 to 2 minutes, beginning with right-sided facial twitching, and associated with labial cyanosis during sleep.
On admission, he had the following measurements: height, 64.0 cm (–0.8 standard deviations); weight, 6485 g (–1.2 standard deviations); chest circumference, 40.0 cm (–1.8 standard deviations); and head circumference, 41.0 cm (–1.1 standard deviations). Failure to thrive, hypotonia, left-sided flaccid paresis, stridor, saddle nose, macroglossia, umbilical hernia, bilateral simian creases, and bilateral fifth fingers shortening and incurved were observed. The skin showed a café-au-lait spot, 3 cm2 in size, to the upper right of the umbilicus; 2 café-au-lait spots, 1 cm2 in size, to the left of the umbilicus; and irregular, streaky areas of hypopigmentation around the umbilicus and on the anterior right chest (Figure 1A). Under ultraviolet (UV) light, irregular, streaky areas of hypopigmentation were observed along the lines of Blaschko on the anterior right chest (Figure 1B).
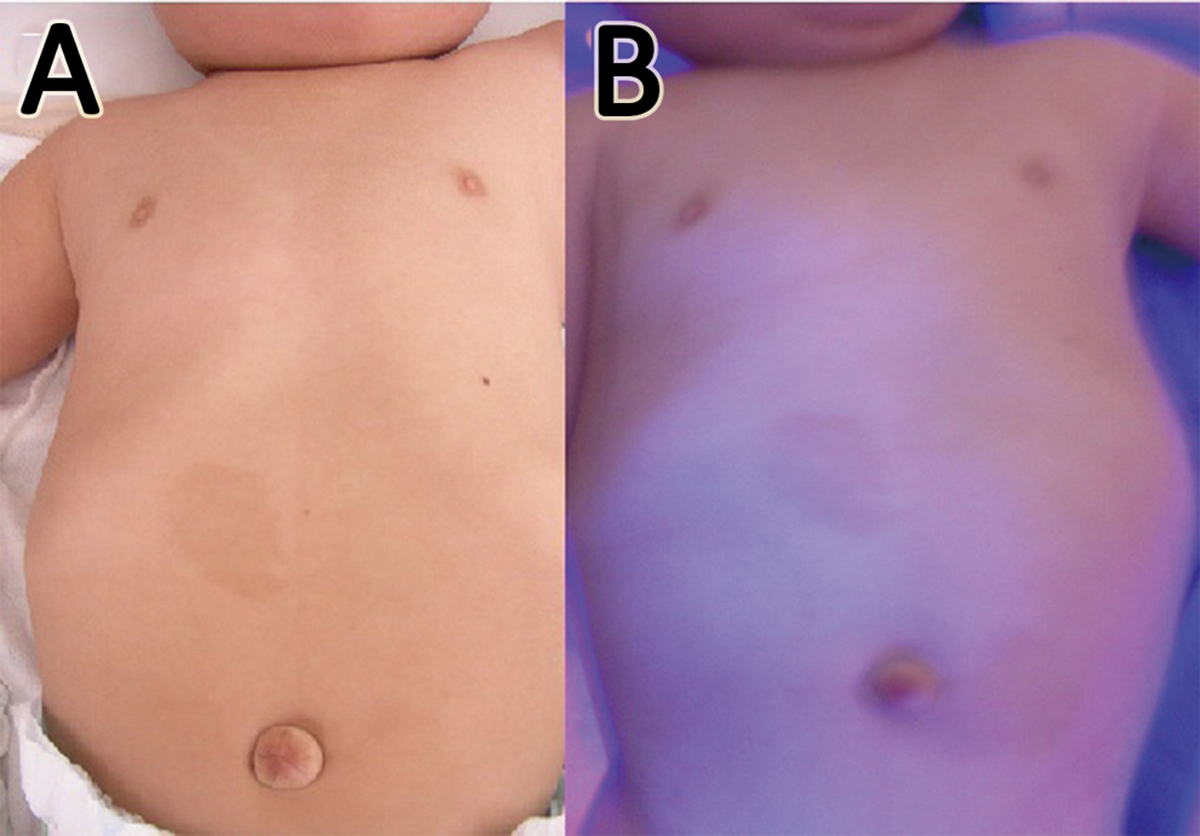
(A) Café-au-lait spots around the umbilicus; and irregular, streaky areas of hypopigmentation on the right anterior chest wall and upper abdomen. (B) Irregular, streaky areas of hypopigmentation along the Blaschko line on the right anterior chest wall and upper abdomen can be seen under UV light.
Laboratory data on admission were as follows: whole blood count (white blood cells, 5810/μL [neutrophils, 28.0%; lymphocytes, 58.0%; monocytes, 2.0%; eosinophils, 3.0%; basophils, 2.0%]; red blood cells, 445 × 104/μL; hemoglobin, 12.6 g/dL; hematocrit, 37.9%; platelets, 30.6 × 104/μL) and serum chemistry (total protein, 6.01 g/dL; albumin, 3.81 g/dL; aspartate aminotransferase, 78.1 IU/L; alanine aminotransferase, 78.3 IU/L; lactate dehydrogenase, 266 IU/L; gamma-glutamyl transferase, 45.9 IU/L; total bilirubin, 0.48 mg/dL; blood urea nitrogen, 7.4 mg/dL; creatinine, 0.28 mg/dL; total cholesterol, 146 mg/dL; glucose, 116 mg/dL; free triiodothyronine, 2.96 pg/mL; free thyroxine, 1.34 ng/dL; thyroid-stimulating hormone, 2.967 IU/mL). All data, except that for liver enzymes, were within normal ranges. The elevation of liver enzymes may have been related to age and medication.
Interictal electroencephalographic (EEG) findings showed frequent spikes, sharp waves, and spike and wave complexes in the right occipital, parietal, and posttemporal regions (Figure 2A). Ictal EEG showed sharp wave bursts in the right-middle temporal region, which developed into polyspikes, and polyspike and wave bursts, involving the right cerebral hemisphere and spreading into both hemispheres (Figure 2B). During the clinical course of his epileptic seizures, he showed neither infantile spasms nor hypsarrhythmia. Brain magnetic resonance imaging (MRI) scans showed focal cortical dysplasia in the right parieto-occipital area, with broad gyri and shallow sulci: axial views show asymmetry due to mild enlargement of the right cerebral hemisphere (Figure 3A), and ipsilateral enlargement of white matter (Figure 3B); and coronal views showing straightening of the right frontal horn (Figure 3C), all of which are characteristic of hemimegalencephaly. 6,7
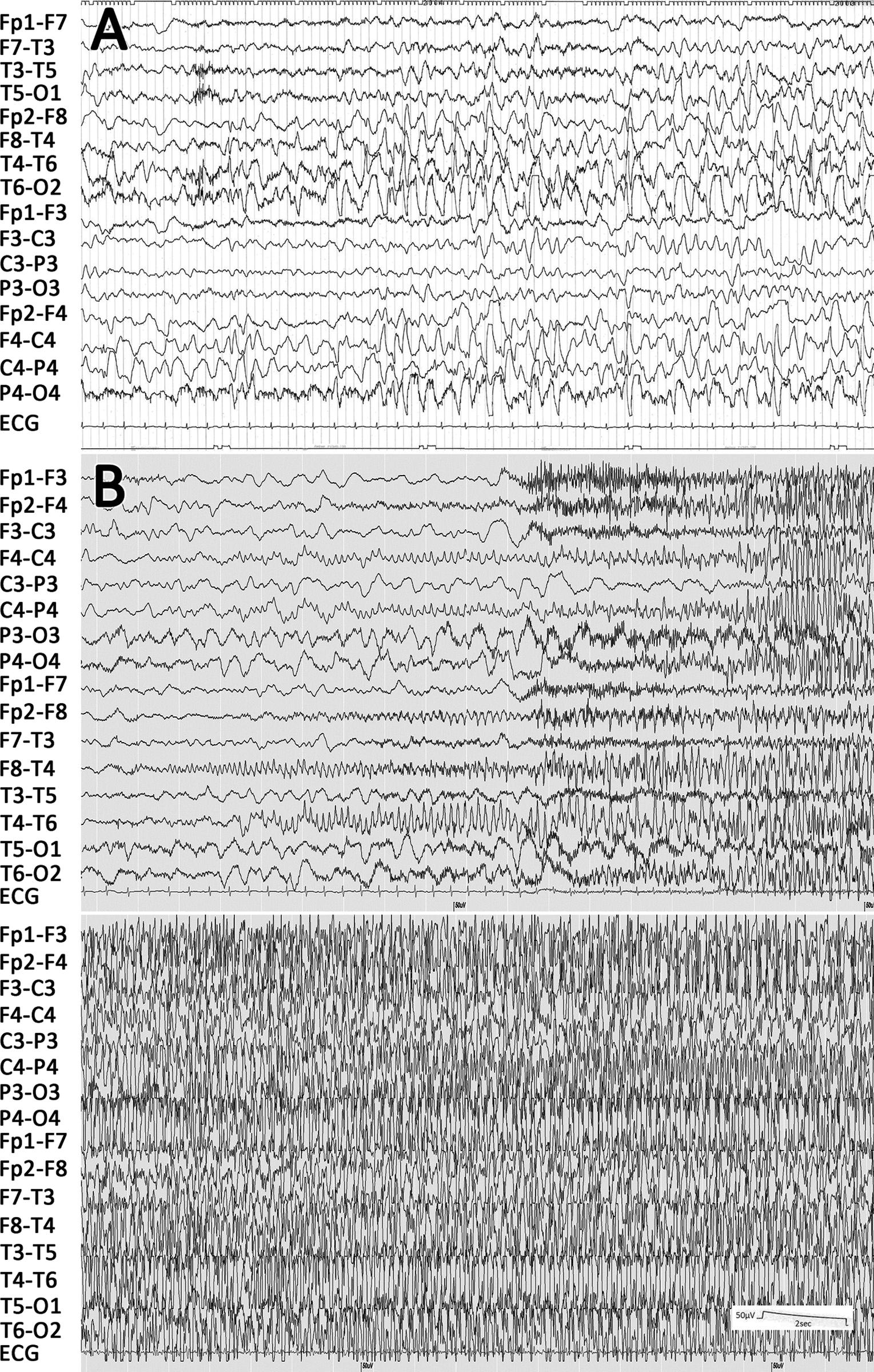
(A) Interictal EEG at age 6 months showing frequent spikes, spike and wave complexes, and high-voltage slow waves in the right occipital and posttemporal regions. (B) Ictal EEG at 6 months of age showing a spike burst in the right-middle temporal region, preceded by right-sided facial twitching, and spreading into the right hemisphere with left unilateral seizures.
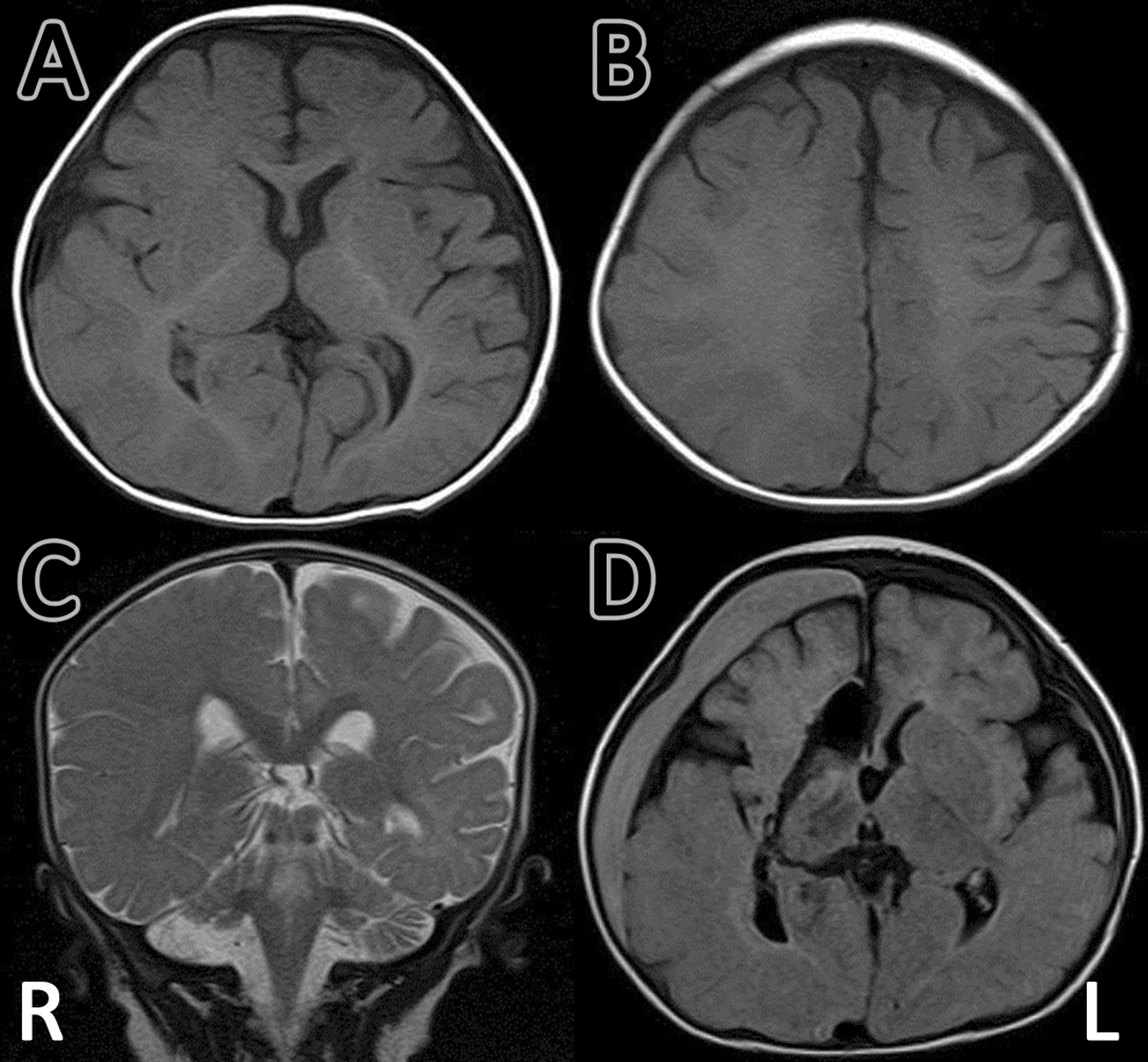
MRI findings before and after neurosurgical intervention (A, B, C). MRI T1-weighted axial image and T2-weighted sagittal image at 5 months of age and before surgery, showing right focal cortical dysplasia in the right parieto-occipital area, with broad gyri and shallow sulci: axial views show (A) asymmetry due to mild enlargement of right cerebral hemisphere and (B) ipsilateral enlargement of white matter; coronal view shows (C) straightening of the right frontal horn, all of which are characteristic of hemimegalencephaly. (D) MRI T1-weighted axial image at 1 year 5 months of age, after surgery. Functional hemispherotomy was performed using the Delalande approach. A subdural hematoma in the right hemisphere resolved spontaneously within 3 months.
Conventional antiepileptic drug therapy, including clonazepam, zonisamide, and clobazam, was ineffective. He had repeated episodes of apnea, hypoxia, cyanosis, and bradycardia, associated with left-sided facial twitching and left-sided hemiconvulsions, and generalized tonic-clonic convulsions. At 10 months of age, functional right-sided hemispherotomy was performed (Figure 3D), without any trial of polytherapy, combination antiepileptic drugs, or alternatives, such as ketogenic diet therapy. After the surgery, his seizures disappeared, but left-sided paresis was observed. At 3 years of age, he is now able to stand and walk with support and is free from epileptic seizures.
Neuropathologic examination of the surgically resected specimen revealed abnormal lamination, high neuronal concentration, columnar structure, dysmorphic neurons, irregular polarity, minute mineralization, CD68+ cells in the cortex (microglia infiltration), balloon cells, heterotopic neurons in the white matter, and many NF+ cells in the cortex and white matter (Figure 4A to F). These neuropathologic findings indicated severe dysplasia, compatible with hemimegalencephaly. 7,8
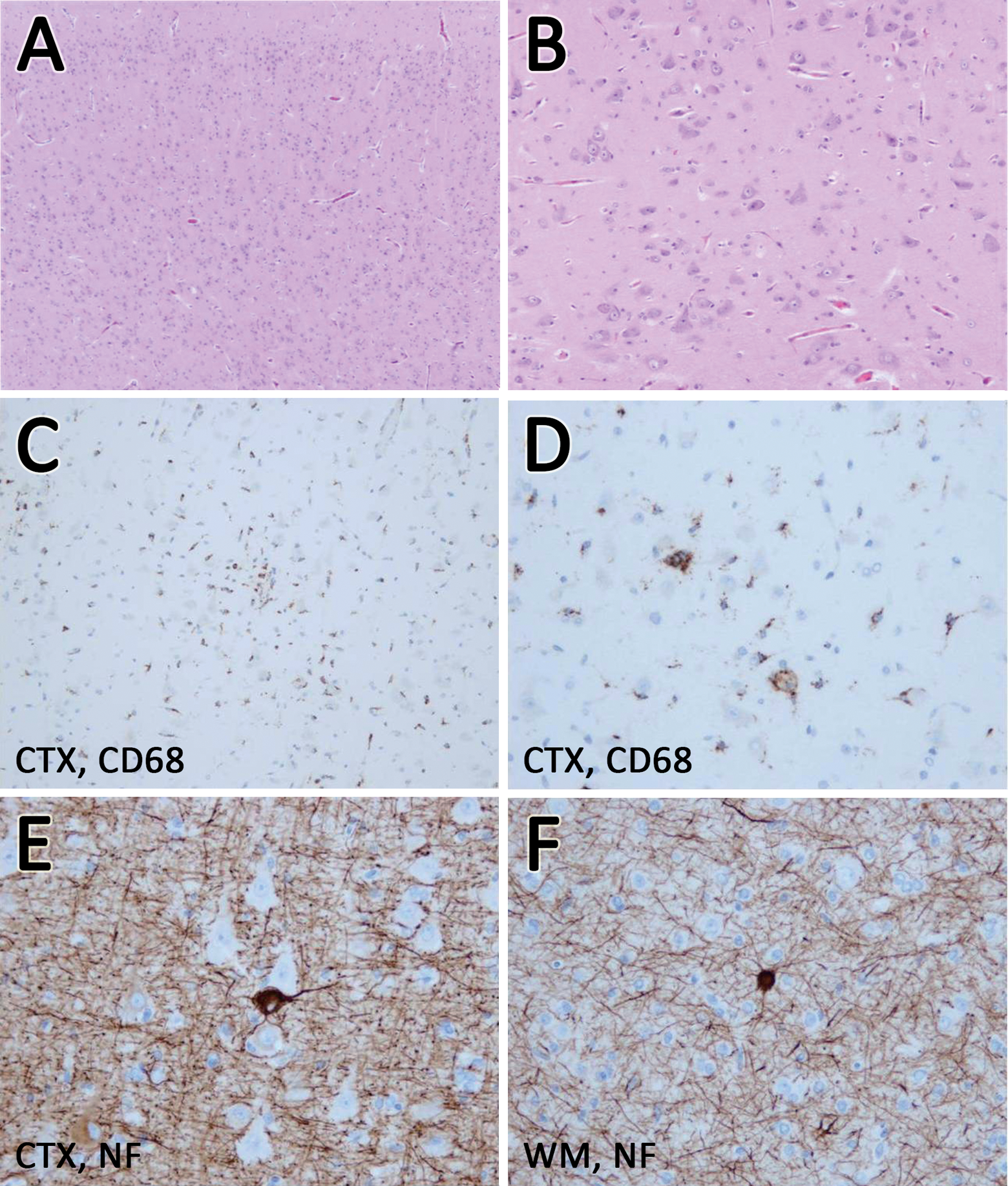
Neuropathologic findings of surgically resected specimen. Abnormal lamination (A); high neuronal concentration (A, B); columnar structure (A, C); dysmorphic neurons (B); irregular polarity (B); minute mineralization (B); CD68+ cells in the cortex (microglia infiltration) (C, D); balloon cells (E); heterotopic neurons and astrocytic gliosis in the white matter (F); and many NF+ cells in the cortex and white matter (E, F). A and B: Hematoxylin-eosin staining; C and D: immunostaining for CD68; E and F: immunostaining for neurofilaments. CTX, cortex; WM, white matter; CD68, cluster of differentiation 68; NF, neurofilament.
Discussion
In this infant with trisomy 21, hypopigmented patches and depigmentation along the lines of Blaschko were observed. This is a characteristic finding of hypomelanosis of Ito. 5,9 –14 The patient developed refractory epilepsy due to hemimegalencephaly, predominantly in the right parieto-occipital area. Although some patients with trisomy 21 suffer from epilepsy and febrile seizures, and develop West syndrome during infancy, 3,4,15 to our knowledge there has been no prior report of refractory epilepsy in a trisomy 21 patient that was caused by either focal cortical dysplasia or hemimegalencephaly.
However, it has been demonstrated that hypomelanosis of Ito can be associated with refractory epilepsy, as well as cortical dysplasia and hemimegalencephaly. 7,16 –19 Hypomelanosis of Ito is caused by a nonheritable mutation. The signs and symptoms of hypomelanosis of Ito include café-au-lait spots, a streaky distribution of hypopigmentation along the lines of Blaschko, intellectual disability, and epilepsy. These skin lesions are clearly visible using a Wood’s lamp. 13 Hypomelanosis of Ito is often associated with chromosomal abnormalities. 5,9 –13,20 Epilepsy in patients with hypomelanosis of Ito is caused by various cortical dysplasia, including hemimegalencephaly. 5 –7,9 –14 Hemimegalencephaly is most often an isolated congenital abnormality, but it is sporadically associated with neurocutaneous syndromes, such as linear nevus sebaceous syndrome, tuberous sclerosis, hypomelanosis of Ito, and, occasionally, neurofibromatosis. 6,21 The striking feature of our present case was the coexisting trisomy 21 and hypomelanosis of Ito, since there is no previous report of these 2 conditions in the same patient. Given the absence of reports describing a combination of trisomy 21 and hemimegalencephaly, it is possible that hypomelanosis of Ito in this case was the underlying clinical condition responsible for hemimegalencephaly. In addition, because hypomelanosis of Ito is often accompanied by chromosomal abnormalities, it may be associated with trisomy 21.
Although treatment of epilepsy was difficult in this case, with conventional antiepileptic drugs being ineffective, the seizures were completely suppressed and the infant’s growth resumed after functional right-sided hemispherotomy. Focal cortical dysplasia and hemimegalencephaly have been seen in several neurocutaneous syndromes, such as hypomelanosis of Ito, neurofibromatosis type 1, linear nevus sebaceous syndrome, and tuberous sclerosis. The clinical presentation in patients with focal cortical dysplasia or hemimegalencephaly usually includes epileptic seizures, which start within the first 6 months of life. The seizures are partial, with secondary generalization, and are often intractable to conventional antiepileptic drugs. Infantile spasms sometimes present in early infancy. Surgical intervention should be considered for patients with symptomatic infantile spasms who have underlying focal cortical dysplasia, usually temporal-parietal-occipital or hemimegalencephaly. Infants with hemimegalencephaly or focal cortical dysplasia can present with multiple daily seizures, developmental stagnation or decline, and hemiparesis. Early surgical intervention, hemispherectomy, or cortical resection provides relief from seizures and improves developmental outcome. 16 –18,22,23 Despite adequate control of his epilepsy, the prognosis for neurologic function is guarded because of the chromosomal abnormality in this patient.
Footnotes
Acknowledgments
The authors would thank the patient’s family for their cooperation.
Author Contributions
TI conceived the main idea and theme for presentation. KO wrote the first draft of the manuscript. AT and TO performed surgical intervention. KO, HM, AT, TO, KA, EN, and TI were involved in clinical management of the patient. MI and HM reviewed the pathologic findings. TI critically reviewed the manuscript and the interpretation.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Informed consent for publication of the case report was given by the patient’s parents.