
Abstract
Childhood leukoencephalopathies are a broad class of diseases, which are extremely rare. The treatment and classification of these disorders are both challenging. Nearly half of children presenting with a leukoencephalopathy remain without a specific diagnosis. Leukoencephalopathy with thalamus and brain stem involvement and high lactate (LTBL) is a newly described childhood leukoencephalopathy caused by mutations in the gene encoding a mitochondrial aminoacyl-tRNA synthetase specific for glutamate, EARS2. Magnetic resonance images show a characteristic leukoencephalopathy with thalamic and brain stem involvement. Here, we report a different clinical course of LTBL supported by typical MRI features in a Turkish patient who presented with a history of failure to walk. The EARS2 gene mutation analysis identified a c.322C>T transition, predicting a p.R108W change. This is the first reported early-onset mild type LTBL caused by a homozygous EARS2 mutation case in the literature.
Keywords
Mitochondrial diseases are a heterogeneous group of disorders resulting from primary dysfunction of the respiratory chain due to both nuclear and mitochondrial DNA mutations. 1 Defective translation of mitochondrial (mit) DNA-encoded proteins, caused by mutations in either the mitochondrial or nuclear genome, often manifest as severe infantile leukoencephalopathy. 2 EARS2 is one of the newest members of nuclear mitochondrial disorders characterized by disturbed mitochondrial translation. 3 This gene encodes the mitochondrial aminoacyl-transfer RNA synthetase specific for glutamate, which is involved in mitochondrial DNA translation.
Leukoencephalopathy with thalamus and brainstem involvement and high lactate (LTBL) was first described in 12 patients, all of whom shared an infantile onset and rapidly progressive disease with severe magnetic resonance imaging (MRI) abnormalities and increased lactate in body fluids. Regarding these 12 patients, phenotyping based on the clinical and MRI features reveal 2 distinct groups: mild and severe. Patients in the mild group could partially recover and regain milestones in the following years whereas the severe group is characterized by clinical stagnation and brain atrophy on MRI. The brain MRI of these patients is hallmarked by extensive symmetrical cerebral white matter abnormalities and symmetrical signal abnormalities of thalamus and brainstem. 4
Since the original publication, we identified 1 patient with early-onset, mild leukoencephalopathy caused by homozygous mutation in EARS2. This new case of a rarely reported disease, which is called LTBL, expands the clinical phenotype of homozygous EARS2 mutations.
Case Report
A 16-month-old girl presented with a developmental delay of gait. She was able to stand with support but could not walk. She was the first child of consanguineous Turkish parents and had no siblings. She was born at term after an uneventful pregnancy and normal delivery. Perinatal period was normal. Weight, height, and head circumference were normal at birth. She achieved head control at 2 months and succeeded in sitting without support at 10 months but could not walk unaided. Neurologic findings were otherwise normal. Laboratory investigations revealed minimal elevations of serum transaminases and creatine phosphokinase (268 U/L, normal <192). At 17 months, these blood parameters returned to normal; however, blood lactate was increased (2.66 mmol/L, normal range 0.5-2.22 mmol/L). Brain MRI at 17 months revealed T2 and fluid-attenuated inversion recovery hyperintensity of the thalami, caudate nuclei, putamen, midbrain, pons, and cerebellar white matter. T2 and fluid-attenuated inversion recovery hyperintensity were observed of periventricular and supraventricular white matter, corpus callosum, and periaquaductal grey matter. Notably, periventricular rim, subcortical U-fibers, internal capsules, and corona radiata were spared and had normal T2 signal (Figures 1 and 2). At age 2.5 years, she manifested a single febrile seizure. Electroencephalography was normal.
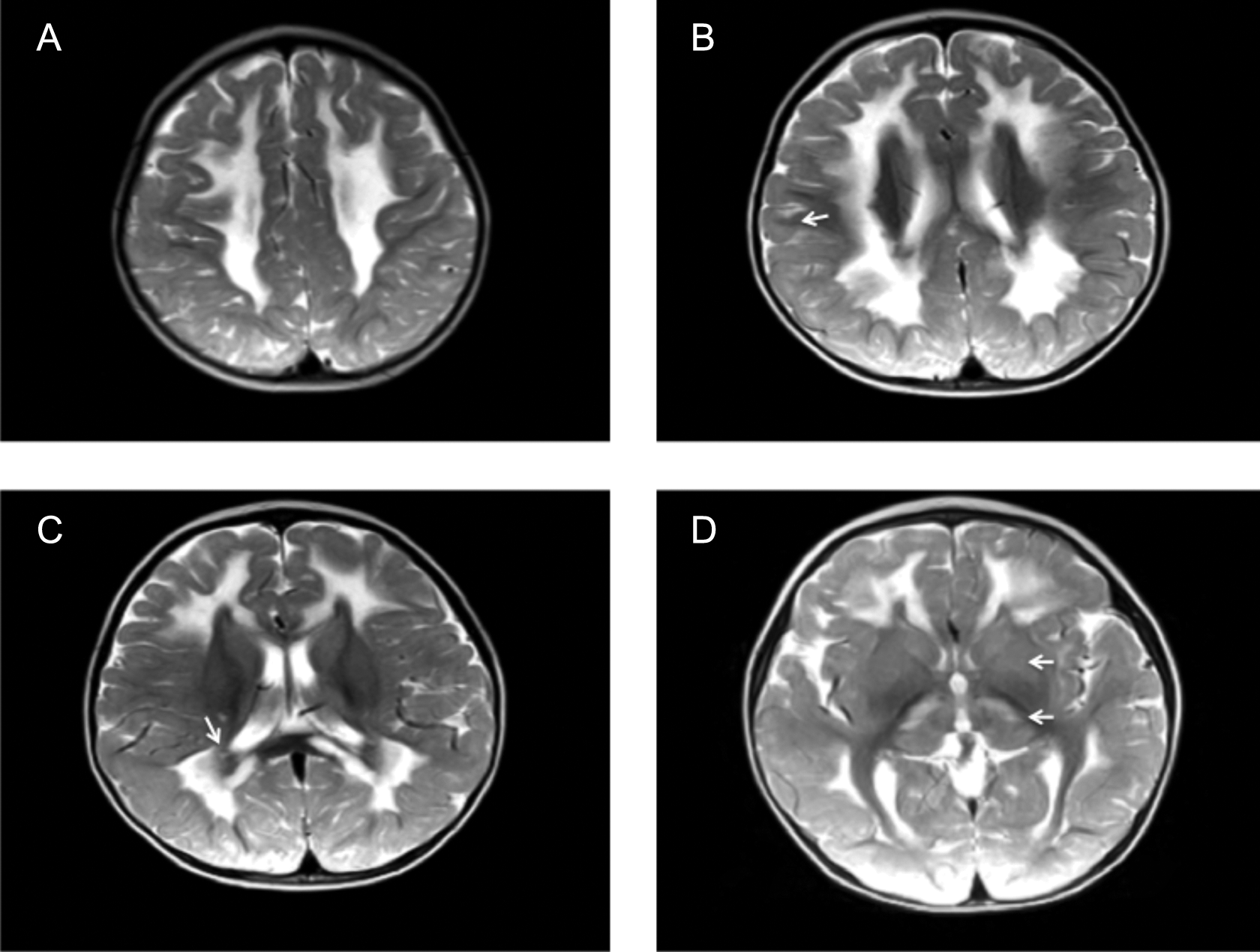
Diffuse abnormal T2 hyperintensity in the cerebral white matter, midbrain, periventricular and supraventricular white matter, corpus callosum and periaquaductal grey matter which spares periventricular rim, subcortical U fibers (B; arrow), periventricular rim (C; arrow), internal capsules and corona radiata (A, B, C). T2 hyperintensity of the thalami, caudate nuclei, and putamen are also demostrated (D; arrows).
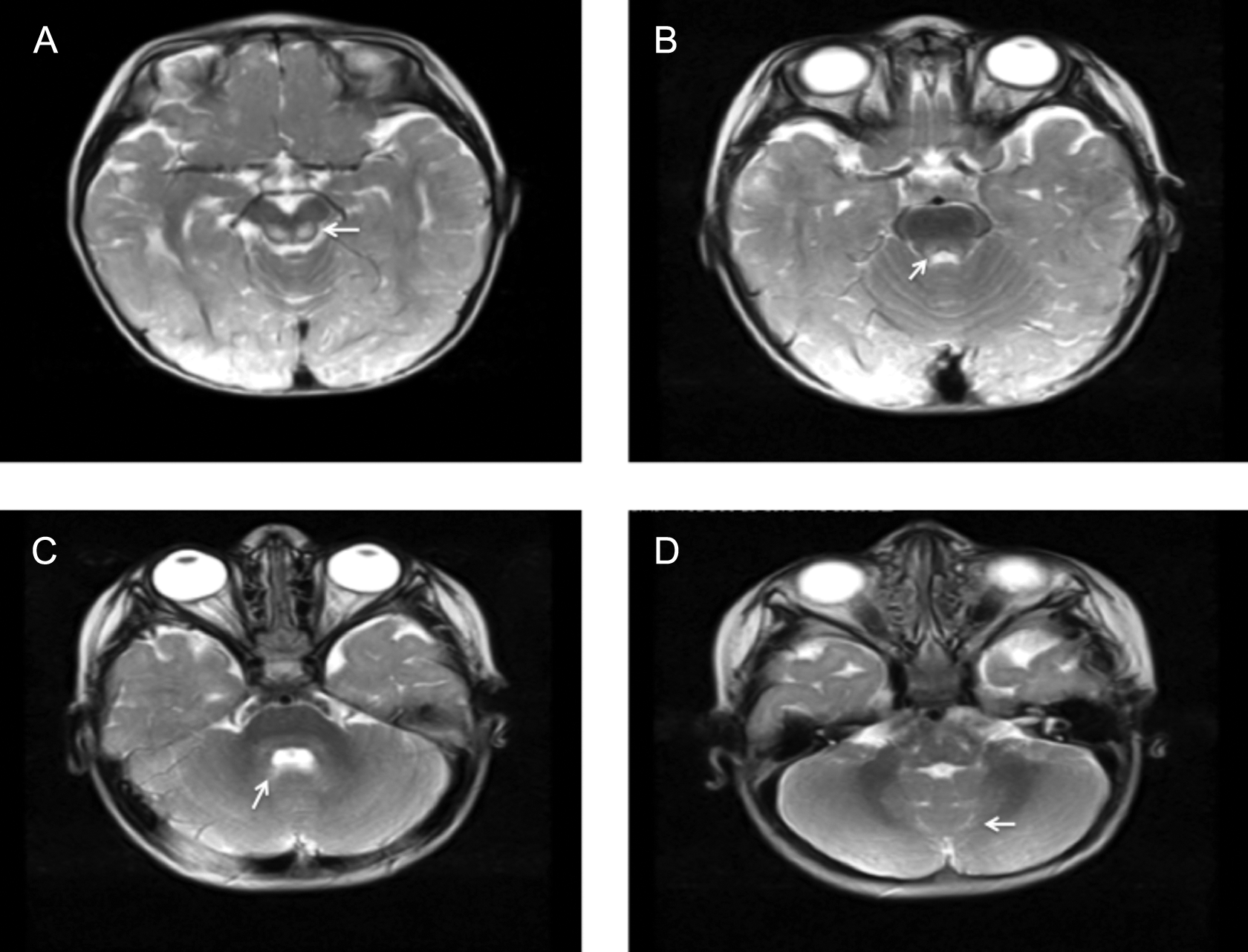
Abnormal T2 hyperintensity in the midbrain (A, B; arrows), pons and cerebellar white matter (C, D; arrows).
Because the MRI pattern and blood lactate value corresponded to those reported in LTBL, after receiving written informed consent from the parents of the patient, we analyzed the gene sequence of EARS2. Gene sequencing of the exons, intron-exon boundaries, and promotor region of the EARS2 gene found homozygous mutations. Both of the maternal and paternal allele harbored a c.322C>T transition, predicting a p.R108 W change. Both parents are heterozygous for the p.R108 W (c.322C>T). To our knowledge, this mutation has previously been reported in heterozygous cases; however, our case presented herein differs from previous reports with being the only homozygous case harboring this mutation in the literature.
Follow-up at 3 years of age reveals independent but ataxic gait. Cognitive abilities are above average. There was no recurrence of seizure of evidence for regression.
Discussion
An LTBL case is presented which is caused by mutations in EARS2 (MIM 612799) gene, encoding a mitochondrial transfer RNA synthetase. Mutations in genes encoding both cytoplasmic and mitochondrial transfer RNA synthetases are of crucial importance which can cause an extensive range of neurological and multisystem disorders. A spectrum of peripheral neuropathy, called Charcot-Marie-Tooth disease (CMT [MIM 613287, 601472, 608323, and 613641]), are caused by damaging mutations encoding cytoplasmic aminoacyl–transfer RNA synthetases including genes AARS (MIM 601065), 5 GARS (MIM 600287), 6 YARS (MIM 603623), 7 and KARS (MIM 601421). 8 Another recessive mutation in DARS2 (MIM 610956), encoding the mitochondria-specific aspartyl–transfer RNA synthetase, causes a disorder called leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL). 8 Furthermore, hypomyelination with brainstem and spinal cord abnormalities and leg spasticity (HBSL, OMIM 615281) is an inherited white matter disorder caused by autosomal recessive mutations in DARS, encoding the cytosolic aspartyl transfer RNA synthetase. 9 The extreme variability and tissue specificity of the diseases caused by mutations in mitochondrial transfer RNA synthetase genes further illustrate the importance of understanding the factors influencing mitochondrial translation in different tissues.
The population prevalence of LTBL is not known, but it is generally considered rare. Most of the LTBL patients harbor compound heterozygous EARS2 mutations. 4 There is one report of homozygous EARS2 mutations, which caused multisystem fatal infantile disease in a consanguineous family of Turkish origin. The proband presented with hypotonia, failure to thrive, and lactic acidosis in the neonatal period. He died of necrotizing bronchopneumonia at 3 months of age. 3 Despite our patient harboring homozygous EARS2 mutation, she presented with a history of failure to walk by age 16 months and an otherwise normal examination. In follow-up, she had independent although ataxic gait. There was no regression in her developmental milestones. This is the first published report of homozygous EARS2 mutations causing mild-type LTBL as far as we are aware.
Initially, in 2012, Steenweg et al 4 identified a series of 12 subjects sharing a set of common features, including (1) mutations in the same single gene, EARS2; (2) similar cardinal neuroimaging features; and (3) a well-defined clinical syndrome. These results provide evidence for the existence of a new specific mitochondrial disease condition: leukoencephalopathy with thalamus and brainstem involvement, and high lactate (LTBL). 4 Our patient’s findings match all 3 criteria.
In conclusion, we present a patient with a mild early-onset leukoencephalopathy with thalamus and brainstem involvement and high lactate (LTBL), hallmarked by consistent MRI features and caused by EARS2 mutations. Abnormality of thalamus and brainstem tracts are MRI findings that point to the correct diagnosis of this newly described neurologic disorder. In combination with consistent clinical features, the findings enable targeting the molecular diagnostics to identify this syndrome.
By this report, we emphasize that a possible mild phenotype of homozygous EARS2 mutations may be underdiagnosed in the population because of relatively mild neurologic symptoms, in contrast to the reported cases with rapidly progressive CNS disease. Therefore, this case widens the clinical spectrum of EARS2 mutations and furthermore shows that a patient with EARS2 mutation homozygosity can develop a mild phenotype.
Footnotes
Author Contributions
BDT wrote the manuscript. BDT, ZSK, EG, KA, SC and KK were involved in the diagnosis. BDT, ZSK, UA and CY were involved in the management of the patient. CY and PLP edited the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Informed consent was taken from the parents for publication of the case report. The Ethics Committee Approval number is: 2015-0032.