
Abstract
Dravet syndrome is a rare and progressive epileptic encephalopathy of infancy. Stiripentol reduces the seizure frequency in patients with Dravet syndrome. We evaluated the clinical characteristics of patients with Dravet syndrome and their response to stiripentol. We retrospectively collected the data of 21 patients (11 females; mean age, 8.2 years, range: 5.4-15 years) with Dravet syndrome who were treated with stiripentol in our outpatient clinic between June 2016 and June 2017. Patients with seizure reduction ≥50% were considered responders. Most of our patients had severe (47%) or moderate (33%) cognitive disabilities, although 14% had mild cognitive disability. There was a significant difference in both status epilepticus and age between the groups with normal/mild versus severe/moderate neurocognitive prognoses. Of the patients, 85.7% were using stiripentol. The mean duration of stiripentol use was 41.2 months (range: 24-64 months). In 12 patients (57%), the seizure frequency decreased by more than 50%, and 2 of them were seizure-free. Status epilepticus was not recorded after stiripentol treatment in 8 of 11 patients with status epilepticus. Despite the small sample size, our results suggest that stiripentol has a favorable efficacy. In addition, considering the absence of status epilepticus after treatment and the negative effects of status epilepticus on cognitive development, early treatment should be initiated in SD patients, for whom disease control is difficult.
Dravet syndrome is a rare progressive epileptic encephalopathy also known as severe myoclonic epilepsy of infancy. 1 Wu et al 2 reported that clinical Dravet syndrome is seen in 1 in 15 700 births, and the estimated incidence of SCN1A-associated Dravet syndrome is 1 in 20 900 births. Dravet syndrome begins toward the middle of the first year of life (range: 2-10 months), usually with febrile, generalized, or unilateral clonic (hemiclonic with alternating sides) long seizures. Later, multiple seizure types occur, including tonic-clonic, myoclonic, focal, and absence seizures. Status epilepticus frequently develops; the course involves somnolence with convulsive or nonconvulsive myoclonus, and fever and certain medications can induce status epilepticus. 1 The main concerns when following these patients are prevention of status epilepticus development and neurocognitive arrest or regression. The prognosis of Dravet syndrome in adulthood depends on the whether the seizures are controlled, which is closely related to patient quality of life. 3,4 The severity of epileptic seizures and frequency of status epilepticus predict the neurocognitive prognosis. 4 Valproic acid, clobazam, bromide, topiramate, and a ketogenic diet are traditional proven therapies in Dravet syndrome. In addition to the above therapeutics, studies involving cannabidiol use are also increasing. 5 Stiripentol belongs to a new class of antiepileptic drugs and has been approved as an add-on therapy to valproic acid and clobazam. In placebo-controlled trials, it reduced the frequency of seizures in patients with Dravet syndrome. 6,7 Stiripentol is structurally unrelated to any other antiepileptic drug, and it acts as an allosteric modulator of the γ-aminobutyric acid (GABA)-A receptor. 8 The efficacy of stiripentol could be related to the potentiation of GABAergic inhibitory neurotransmission and enhancement of the action of benzodiazepines. 8,9
This study evaluated both the clinical characteristics and response to stiripentol of patients with Dravet syndrome who were followed in our clinic.
Methods
We retrospectively collected the data of 21 patients with Dravet syndrome who were examined in our outpatient clinic between June 2016 and June 2017. Patient selection and diagnosis followed the diagnostic criteria for Dravet syndrome of the International League against Epilepsy classification scheme. 10
The patients were tested within the previous year. Patients were classified as normal or having mild, moderate, or severe intellectual disability according to the results of psychometric tests routinely used to evaluate cognitive function in patients with refractory epilepsy. In total, 3 of the 21 patients were younger than 72 months of age and were evaluated using the Denver II test; the remaining 18 patients were tested with the Wechsler Intelligence Scale for Children–Revised (WISC-R). According to their WISC-R total IQ scores, the 18 patients older than 72 months were divided into 4 groups: (1) normal, IQ >85; (2) mild disability, IQ 71 to 84; (3) moderate disability, IQ 55 to 70; and (4) severe disability, IQ < 55.
The patients were subject to a neurologic examination, and cognitive function tests and treatment compliance was evaluated. One patient was excluded because of treatment incompatibility and another because there was no information on the seizure frequency or psychometric test results.
Demographic characteristics were recorded, including age at seizure onset, sex, type and duration of seizures, family history of febrile seizures and epilepsy, history of status epilepticus, age at the time of the most recent status epilepticus, electroencephalographic (EEG) findings (at baseline and most recent), the frequency of seizures, psychometric testing, and all antiepileptic drugs used.
We classified seizures as tonic-clonic, myoclonic, clonic, or atypical absence. Seizure frequency was assessed via parent diaries and the records of medical staff. The last 12 weeks before stiripentol administration was defined as baseline, and each subject had at least 12 weeks of baseline data. The frequency of seizures was classified as daily (>4/wk), weekly (>3/mo), or monthly (1-3/mo). Group 1 included patients with a more than 50% decrease in the seizure frequency (responsive), whereas group 2 included patients with a less than 49% reduction, or no change, in seizure frequency (unresponsive to treatment). Seizure remission was defined as being free from seizures for at least 1 year at the last visit (Table 1).
Summary of Stiripentol Effectiveness.
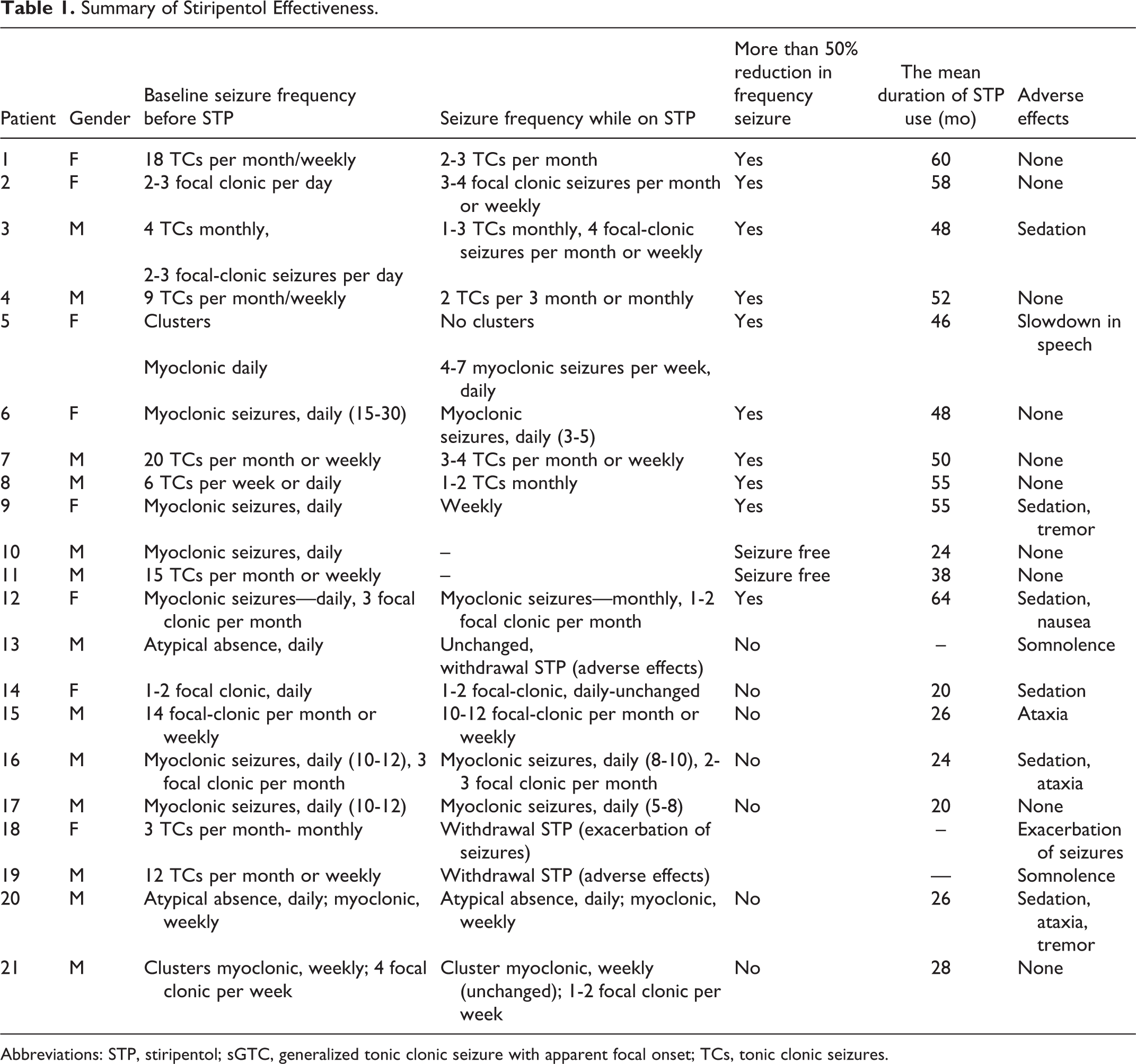
Abbreviations: STP, stiripentol; sGTC, generalized tonic clonic seizure with apparent focal onset; TCs, tonic clonic seizures.
The study was reviewed and approved by the Institutional Ethics Committee of the Istanbul Faculty of Medicine. All procedures performed in the study were in accordance with the ethical standards of the institutional and national research committees and the 1964 Declaration of Helsinki and its amendments or comparable ethical standards. All participants or their legal representatives provided written informed consent.
Statistical Analysis
The statistical analysis was performed using SPSS for Windows software (version 21.0; IBM Corp, Armonk, NY). The normality of the data was analyzed using the Shapiro-Wilk test and histograms. Categorical variables were compared among the groups using the chi-square (χ2) test and Fisher exact test in the case of an expected cell size <5. Student t test was used to compare normally distributed continuous variables, whereas the Mann-Whitney U test was used for nonnormally distributed continuous variables. A 2-tailed P value of <.05 was considered statistically significant.
Results
Patient Population
The 21 patients comprised 11 girls and 10 boys. Their mean age was 8.2 years (range: 5.4-15 years). The mean age at seizure onset was 4 months (range: 3-12 months). Two of our patients had SCN1B mutations and the other 19 had SCN1A mutations. Fever triggered the first seizure in 85% of the patients (n = 18), while convulsive status epilepticus was the initial seizure in 52% (n = 11). There was a family history of febrile seizures in only 14% of the patients (n = 3). The medications used by the patients at the last visit included levetiracetam (n = 3, 14.2%), topiramate (n = 6, 25.8%), clobazam (n = 15, 71%), valproic acid (n = 14, 66.7%), and stiripentol (n = 18, 85%). During the study, 3 (14.2%), 13 (62%), and 5 (23.8%) patients were on combination treatment with 2, 3, and 4 antiepileptic drugs, respectively.
Based on the Denver II test, 1 of 3 patients was evaluated as normal, that is, no delay in any domain, whereas 2 patients had 1- and 1.5-year delays in language development only, who were classified as having mild cognitive disability by the blinded test administrator. Ten (47%) patients had severe, and 7 (33%) patients had moderate cognitive disability; 3 (14%) other patients had mild cognitive disability.
There was no significant difference in sex, type of seizure, or response to stiripentol between the groups with normal/mild and severe/moderate neurocognitive outcomes. However, there were significant differences in both status epilepticus and age between the 2 groups (P = .03 and P = .04, respectively) (Table 2).
Variables That Predict Neurocognitive Outcomes
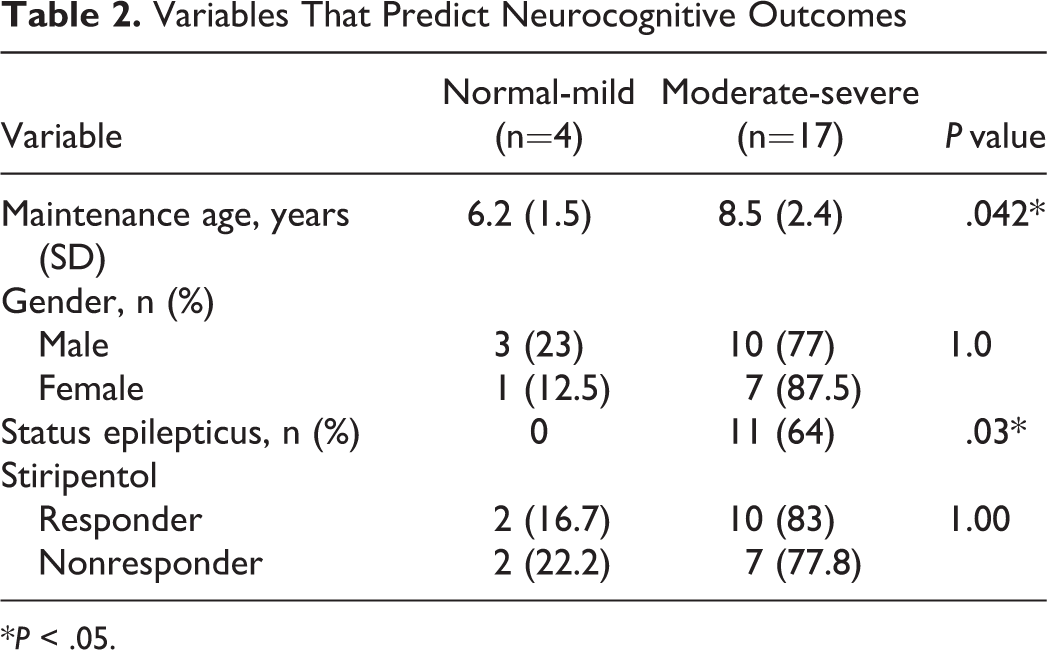
*P < .05.
Seizures
Clinical seizures were recorded in 90% of our patients within the previous 3 months. The families of 2 seizure-free patients had not documented any seizures for 20 months (6-year-old patient) and 30 months (7-year-old patient). Both of these patients were using 3 antiepileptic drugs (topiramate, clobazam, and stiripentol and topiramate, valproic acid, and stiripentol). Seizures occurred daily in 28.5% of the patients (n = 6), weekly (>3/mo) in 38% (n = 8), and monthly (1–3/mo) in 23.8% (n = 5). Table 1 shows the reduction in seizure frequencies from baseline.
The predominant seizure types were tonic–clonic in 33.3% of the patients (n = 7), clonic in 38% (n = 8), myoclonic in 19% (n = 4), and atypical absence in 10% (n = 2).
All patients had interictal EEG records. Only 1 patient (the youngest) had a normal EEG; 11 patients showed multifocal generalized multiple spikes and multiple spike slow waves, 6 showed generalized spikes, and 3 had focal slow spike wave activity.
Stiripentol Response
Of the patients, 85.7% (n = 18) were using stiripentol. Stiripentol was halted in 2 patients due to excessive somnolence (at weeks 2 and 3, respectively) and in 1 patient due to increased seizure frequency. The mean duration of stiripentol use was 41.2 months (range: 24 to 64 months), whereas the mean maximum tolerated dose was 33.2 mg/kg/d (range: 31 to 40 mg/kg/d). In 12 patients (57%), the seizure frequency decreased by more than 50%, and 2 of them were seizure-free. One of the 2 seizure-free patients was 7 years old and had been using stiripentol for 38 months, and the family did not record a clinical seizure for 30 months. The other patient was 6 years old and had been using stiripentol for 24 months, and the family did not record a seizure for 20 months.
The mean time between the first seizure and initiation of stiripentol did not differ statistically between the responsive and unresponsive patients, being 47.1 months and 47.5 months, respectively. Before beginning treatment with stiripentol, 11 patients (52%) had a history of status epilepticus. In 8 of these patients, no status epilepticus episodes were observed from initiation of stiripentol to the last visit (mean, 44 months [range: 37 to 64 months]).
When the patients who responded to stiripentol treatment were compared with those who did not benefit from the treatment, there was no significant group difference in seizure type, sex, or history of status epilepticus (Table 3).
Patient Characteristics According to Response to STP Treatment at the Last Follow-up Visit.
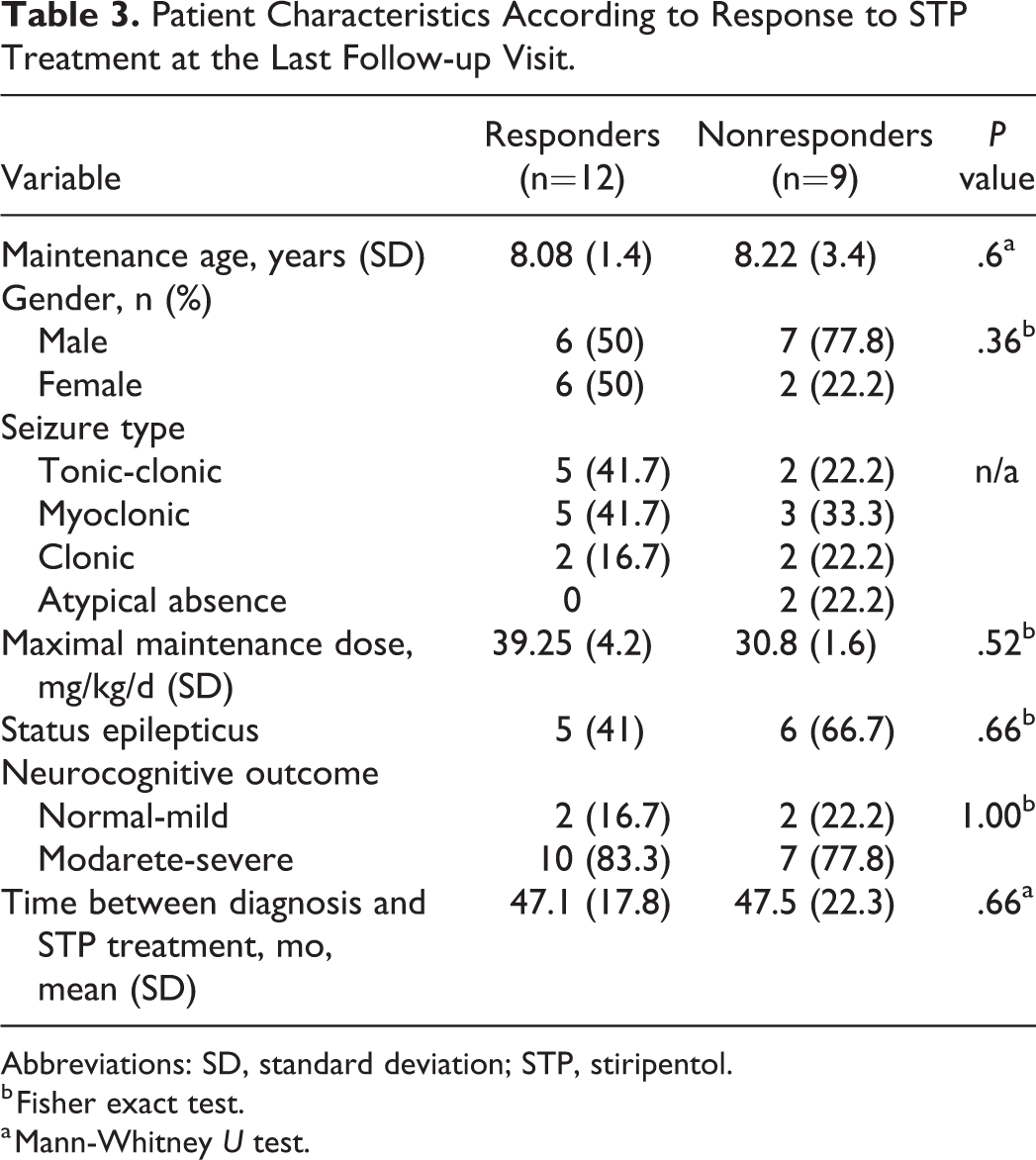
Abbreviations: SD, standard deviation; STP, stiripentol.
b Fisher exact test.
a Mann-Whitney U test.
Adverse effects were reported in 8 patients, with the most common being sedation (6/8) and ataxia (5/8). Other reported side effects included tremor, speech slowdown, and nausea (Table 1). In addition, stiripentol was disrupted in 2 patients because of severe somnolence and in a patient because of increase in seizure number. No patient had laboratory anomalies in liver, renal function, or hematologic parameters.
Discussion
This study evaluated patients with Dravet syndrome who were examined in our clinic in the past year, had detailed records of the neurocognitive outcome, and received stiripentol. In our study group, the male-to-female ratio was 1:1, similar to recent studies. 11 -13 In Dravet syndrome, there is a family history of epilepsy or febrile seizures in less than 25% of cases. 13,14 Similarly, 14% of our patients had a family history. In addition, fever triggered the first seizure in the majority of our patients (85%), consistent with the literature. 13 -15
Status epilepticus prevention is one of the major problems in Dravet syndrome. Although the reported effect of status epilepticus on prognosis is unclear, 11,16 some studies suggest that seizure control at an early age has a positive effect on both seizures and the cognitive prognosis. 17 We found a strong relationship between status epilepticus history and cognitive prognosis. In addition, the normal/mild group was 2.5 years younger than the moderate/severe group. As cognitive deterioration occurs with age in Dravet syndrome, it is noteworthy that 4 patients in the normal/mild group had no history of status epilepticus. In comparison, 11 of 17 patients in the moderate/severe group had histories of status epilepticus and almost half of them (5/11) required one or more hospitalizations within a month; this suggests that status epilepticus is a strong predictor of neurocognitive prognosis. One limitation of this study was the short follow-up duration. In addition, the study design was not prospective, so that we could not prove a relationship between seizures/status epilepticus episodes and cognitive deterioration. We did not observe status epilepticus in 8 of the 11 patients with a history of status epilepticus after stiripentol was started. Therefore, we believe that initiating stiripentol early is of utmost importance to prevent the development of more frequent seizures and status epilepticus, and to reduce the associated cognitive retardation. Unfortunately, in our subjects, the interval from initial diagnosis to the start of stiripentol treatment was quite long because stiripentol, which costs about 400 USD monthly, was first licensed in Turkey in 2012.
Despite current treatment, the seizures persisted in 90% of our patients and 28.5% had daily seizures. Complete seizure control is very difficult to achieve in Dravet syndrome. Our institution is a reference center, which may have resulted in our seeing an epilepsy population that was severe and resistant. A randomized placebo-controlled study of patients with Dravet syndrome found that add-on stiripentol to clobazam and valproic acid treatment caused a >50% reduction in seizure frequency in 71% of patients. 6 In a prospective, open-label, multicenter study conducted in Japan, after a 56-week follow-up period with stiripentol treatment, the seizure frequency decreased by more than 50% in 54% of patients. 16 In our study group, after a longer follow-up period (mean, 42 months) with stiripentol treatment, the seizure frequency decreased by more than 50% in 57% of the patients, and 2 of them were seizure-free.
In patients with Dravet syndrome, the clinical findings, frequency, and severity of seizures may change during follow-up, and different types of seizures may occur. Therefore, it is very difficult to identify the type of seizure for which stiripentol is most effective. Both retrospective and prospective studies have addressed tonic-clonic and clonic seizures, whereas myoclonic seizures have been ignored. 6,18 In our small study cohort, we were unable to find a relationship between the response to stiripentol and predominant type of seizure.
Previous studies have shown that interictal EEG abnormalities increase with age in Dravet syndrome. 13,15 Similarly, there were no normal interictal EEG records among our study group, in which the youngest patient was 5.4 years old.
In conclusion, despite the small sample size, our results suggest that stiripentol has a favorable efficacy and safety profile in Dravet syndrome. Considering the low rate of status epilepticus after treatment and the negative effect of status epilepticus on cognitive development, early initiation of stiripentol could have a favorable effect in this patient population, in whom disease control is difficult.
Footnotes
Author Contributions
All authors made a substantial contribution to the concept and design of the work; and acquisition, analysis and interpretation of data. Drafted the article. Approved the version to be published.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Local ethical committee of Istanbul Medical Faculty approved this study (2018/263).