
Abstract
Aspartylglucosaminuria (AGU) is a recessively inherited neurodegenerative lysosomal storage disease characterized by progressive intellectual disability, skeletal abnormalities, connective tissue overgrowth, gait disturbance, and seizures followed by premature death. AGU is caused by pathogenic variants in the aspartylglucosaminidase (AGA) gene, leading to glycoasparagine accumulation and cellular dysfunction. Although more prevalent in the Finnish population, more than 30 AGA variants have been identified worldwide. Owing to its rarity, AGU may be largely underdiagnosed. Recognition of the following early clinical features may aid in AGU diagnosis: developmental delays, hyperactivity, early growth spurt, inguinal and abdominal hernias, clumsiness, characteristic facial features, recurring upper respiratory and ear infections, tonsillectomy, multiple sets of tympanostomy tube placement, and sleep problems. Although no curative therapies currently exist, early diagnosis may provide benefit through the provision of anticipatory guidance, management of expectations, early interventions, and prophylaxis; it will also be crucial for increased clinical benefits of future AGU disease-modifying therapies.
Aspartylglucosaminuria (AGU) is a progressive neurodegenerative lysosomal storage disease characterized by developmental delay, intellectual disability, skeletal abnormalities, connective tissue overgrowth, gait disturbance, and ultimately premature death. AGU is caused by biallelic pathogenic variants in the AGA gene on chromosome 4, which encodes aspartylglucosaminidase (
Aspartylglucosaminidase deficiency and subsequent accumulation of undegraded aspartylglucosamine and other glycoasparagines leads to vacuolization of lysosomes and cellular dysfunction. Clinically, this will manifest as developmental delays and regression, frequent bacterial infections, and connective tissue dysfunction. Patients with AGU often die prematurely from bacterial infections, primarily pulmonary infections. 1
The objective of this review is to describe the clinical presentation of AGU, its current symptomatic management, and present unmet needs. We hypothesize that delayed diagnosis and underdiagnosis (ie, autism secondary to AGU) are frequent, especially in the non-Finnish population, and result in a greater burden to both the patient and caregiver.
Disease Overview
Pathogenesis
AGU is a recessively inherited disease that affects people worldwide of all ethnic groups although more frequently identified in the Finnish population. 2 Because of a founder effect, up to 98% of disease alleles in Finland have the AGUFin major variant (2 nucleotide changes: G482A and G488C). 2 -4 A second variant, AGUFin minor variant (deletion of 2 base pairs within exon 2) results in premature translation termination and accounts for 1.5% of disease alleles in Finland. 5 Both pathogenic variants result in decreased aspartylglucosaminidase activity. 6
The pathogenesis of AGU is due to defects in glycoprotein degradation. In glycoproteins with asparagine-linked sugars, there is an N-glycosidic bond between the carbohydrates and proteins. 7 Aspartylglucosaminidase is responsible for breaking this N-glycosidic bond within the lysosome, so that the carbohydrate and protein moieties may be further degraded to free sugars and amino acids by the respective enzymatic pathways. 7 Consequently, in AGU in which this enzyme is deficient or dysfunctional, there is an accumulation of glycosaparagines, which can be toxic to cellular function over time. 7
Worldwide, more than 30 AGA variants have been identified in patients with diverse ethnicities (Table 1). Of these AGA variants, approximately 50% are missense mutations (eg, c.439T>C [p.S147P] in a Qatari patient 8 ) with the remaining mutations consisting of microdeletions (eg, Del T787 in a patient from Mauritania 9 ) and insertions (eg, 7-nucleotide insertion between exons 3 and 4 in a Japanese patient 10 -12 ) as well as splice mutations, gross deletions, and complex rearrangements (eg, Del 807-940, resulting in the exclusion of exon 8 in an African American patient 13,14 ). One missense mutation, c.302C>T (p.A101 V), was detected in an AGU patient from Italy and another from England. 15 In addition, c.214T>C (p.S72P) was identified in 8 Palestinian AGU patients from 3 unrelated families. 16,17 Furthermore, in one Mexican/Italian patient, only neutral polymorphisms were detected, suggesting that the variant lies within the noncoding or regulatory regions of the AGA gene. 15 The clinical presentation of these sporadic variants will be discussed further in a subsequent section.
Sporadic AGA Variants Identified in AGU Patients Worldwide.
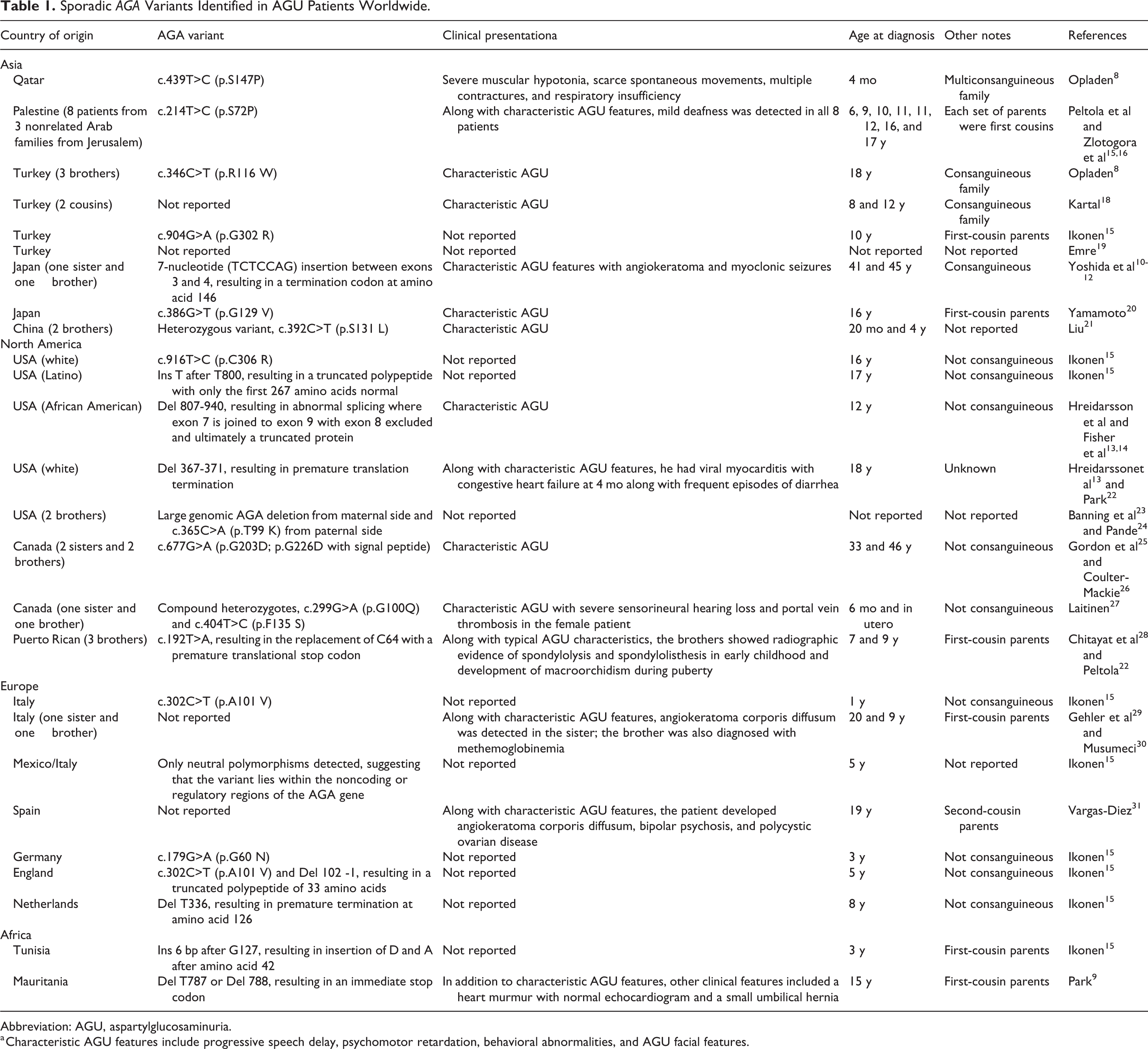
Abbreviation: AGU, aspartylglucosaminuria.
a Characteristic AGU features include progressive speech delay, psychomotor retardation, behavioral abnormalities, and AGU facial features.
Symptomatology/Presentation
AGU is a progressive disease that impacts the entire body and leads to intellectual disability, autistic features, and abnormal skeletal and connective tissue growth with gait abnormalities. Birth measurements in AGU patients are typically normal, though an early infantile growth spurt has been described. 32 Developmental delays, such as delayed speech and clumsy walking, around 12-15 months of age are often the first neurologic signs of AGU. Children tend to have macrocephaly, abdominal and inguinal hernias, as well as recurrent respiratory and ear infections leading to multiple sets of tympanostomy tubes and early tonsillectomy/adenoidectomy. 1 AGU patients experience progressive intellectual disability that becomes more pronounced throughout life; it is mild in elementary school–aged children (Intelligence Quotient [IQ] score of 50-70), progressing to moderate disability (IQ score of 35-49) in teenagers and young adults characterized by loss of reached skills and with further deterioration in adults resulting in severe/profound intellectual decline (IQ score ranging from less than 20 to 34). 33 In a more recent publication, cognitive impairment was more severe and did not correlate with age in a cohort of 7-14-year-old Finnish AGU patients. 34 In this cohort, the full-scale IQ could only be calculated in 5 of 21 patients and ranged from 41 to 52. 34
Similarly, socialization (as measured by ability to ask for assistance, work alone for 20 minutes, and identify emotions), self-help (eg, independent feeding, dressing, using the bathroom, and walking near home), language (ability to state their name and describe immediate and daily experiences), and motor skills (eg, independent walking, biking, skiing, and skipping) follow a functional decline with age while behavioral problems worsen. 35 AGU patients can also have autistic features with reduced social interactions, deficiencies in communication skills (eg, lack of understanding gestures, social cues, or facial features), as well as sensory abnormalities. Behavioral disturbances, including tantrums, violent behavior, hyperactivity, and withdrawal, are also commonly observed in AGU patients. 35 In addition, disruptive sleep patterns affect up to 58% of individuals with AGU; in children, settling difficulties were more commonly reported, while adults tend to struggle with fragmented sleep patterns. 36
AGU is also characterized by skeletal and connective tissue abnormalities. Increased head circumference, characteristic facial features (eg, macroglossia, thick lips, low and wide nasal bridge, short nose, puffy eyelids, and broad face), and oral mucosal overgrowth 37 are observed early and become more prominent with age. Although children with AGU are often tall because of an early growth spurt, adult stature is usually below normal because of attenuated pubertal growth. 38 Skeletal deformations may be observed in some patients, including misshapen ribs, thin long bones, and vertebral abnormalities as well as gait impairment. 1 Some patients develop scoliosis, and others can have an unusually short neck or strong valgus position in their knees. In addition, the ocular lens may develop crystalline deposits. Auto-fluorescent inclusion bodies and thickened retinal nerve fiber layer were described in a pair of brothers with AGU although clinical significance of this finding is unclear. 39
Though they have early macrocephaly, AGU patients experience a reduction in head size that may be accompanied by a thickened skull, suggesting a reduction in brain volume. 40 Brain magnetic resonance image (MRI) analysis showed reductions in subcortical gray matter structures in an AGU patient compared to their unaffected twin. 41 Additional studies in patients with AGU identified T2 hypointensities within the thalami, especially the pulvinar nuclei, beginning at 3.5 years. 18,42 White matter abnormalities are also described, 8,20 including increased T2 signal in the periventricular white matter, poor gray-white matter differentiation, as well as cerebral and cerebellar atrophy associated with ventricular dilatation. 43 The clinical manifestations of these imaging abnormalities are not well-understood; however, it is apparent that individuals with AGU have progressive neurologic disease. In addition to the progressive behavioral and cognitive decline with age, up to 28% of adults with AGU reported having epileptic seizures. 44 Although not commonly reported, cases of epileptic seizures have also been reported in children. 44 Cases of startle epilepsy 45 and sleep-related epileptic seizures have also been reported. 46
Recurring diarrhea as well as respiratory and ear infections are recognized as early symptoms in AGU patients, occurring as early as <2 years of age. 1 Furthermore, up to 7% of female and 4% of male AGU patients suffer from chronic arthritis, which begins in childhood 47 ; carriers are also predisposed to developing chronic arthritis. 48
Some AGU patients may have clinical presentations that differ from the characteristic AGU presentation, which may be due to the presence of other autosomal recessive diseases due to consanguinity in many patients (Table 1). For example, a Qatari AGU patient presented with severe muscular hypotonia, scarce spontaneous movements, multiple contractures, and respiratory insufficiency, 8 whereas hearing loss was detected in 8 Palestinian patients from 3 unrelated families 16,17 as well as 1 Canadian AGU patient. 27 In addition, angiokeratoma and myoclonic seizures were observed in a Japanese patient, 10 -12 hepatosplenomegaly in 2 Chinese patients, 21 viral myocarditis with congestive heart failure in a white patient from the United States, 13,22 macroorchidism in 3 Puerto Rican patients, 28,49 and angiokeratoma corporis diffusum in an Italian patient 29,30 and a Spanish patient 31 have been observed.
Path to Diagnosis
Genetic Testing
Low global awareness of AGU may result in delayed diagnosis of AGU, or patients may initially be diagnosed with autism spectrum disorder before receiving the diagnosis of AGU. If AGU is suspected, genetic testing is required for a definitive diagnosis of AGU. Current genetic tests, which can be ordered by a doctor or other qualified health care professional, analyze the full AGA gene using blood or saliva samples. Deletions involving the AGA gene may be found on chromosomal microarray testing, while sequence variants may be identified by next generation or Sanger sequencing platforms, such as commercially available genetic panels or whole exome sequencing. Both academic as well as commercial laboratories offer genetic testing for AGU.
Biochemical Diagnosis
Although genetic testing is required for a definitive AGU diagnosis, biochemical tests on the basis of accumulated aspartylglucosamine in urine are useful for screening populations. 50,51 Subsequent enzyme tests can confirm the lack of aspartylglucosaminidase activity in serum, plasma, or lymphocyte samples. 52
AGU may also be detected prior to birth using biochemical tests. However, biochemical detection of glycoasparagines in amniotic fluid may not reliably detect AGU, 53 necessitating the culturing of amniotic fluid cells for aspartylglucosaminidase activity 54 or genetic testing for prenatal AGU detection. Absence of aspartylglucosaminidase activity in an umbilical cord serum sample from a newborn has also been demonstrated. 55
Diagnostic Characteristics of AGU
Table 2 describes the classical clinical features of AGU at various ages; Figures 1 to 3 show changes in the facial features of AGU patients over time. Although many features are nonspecific, identification of the early clinical features of AGU may aid in its diagnosis at a younger age.
Features of AGU Occurring From Early Childhood to Adulthood.
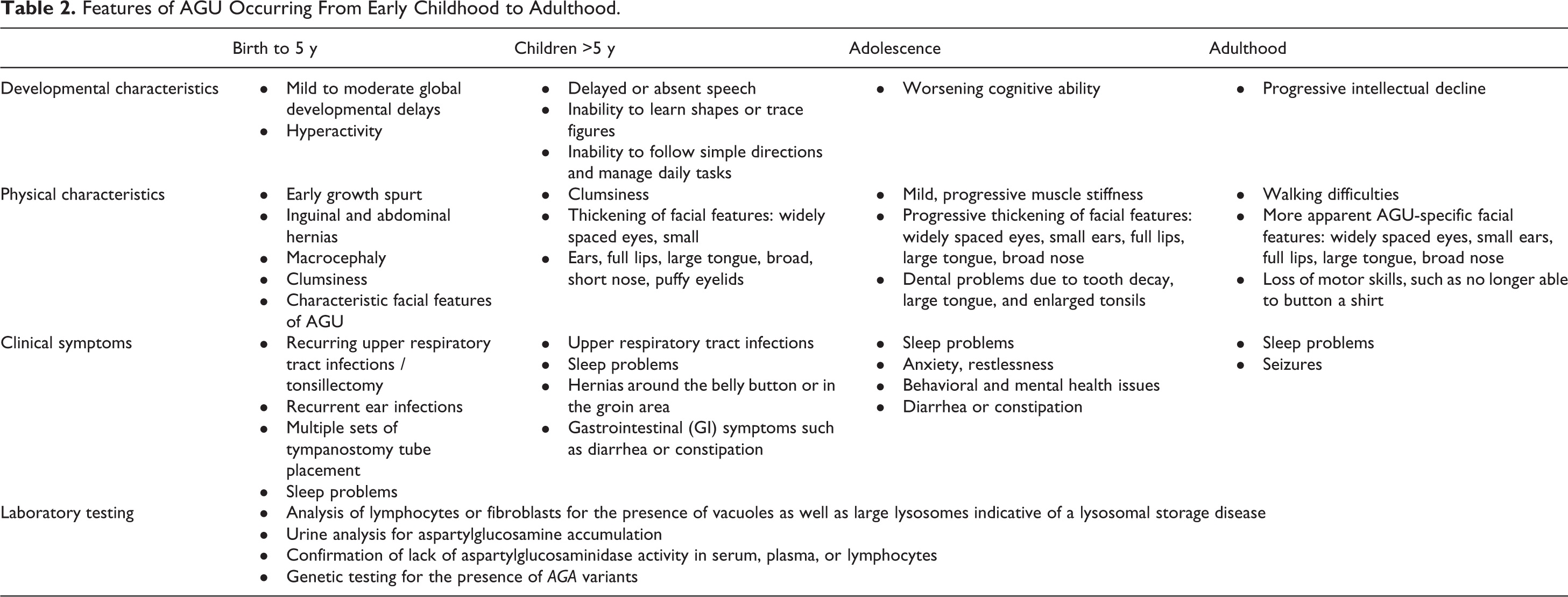
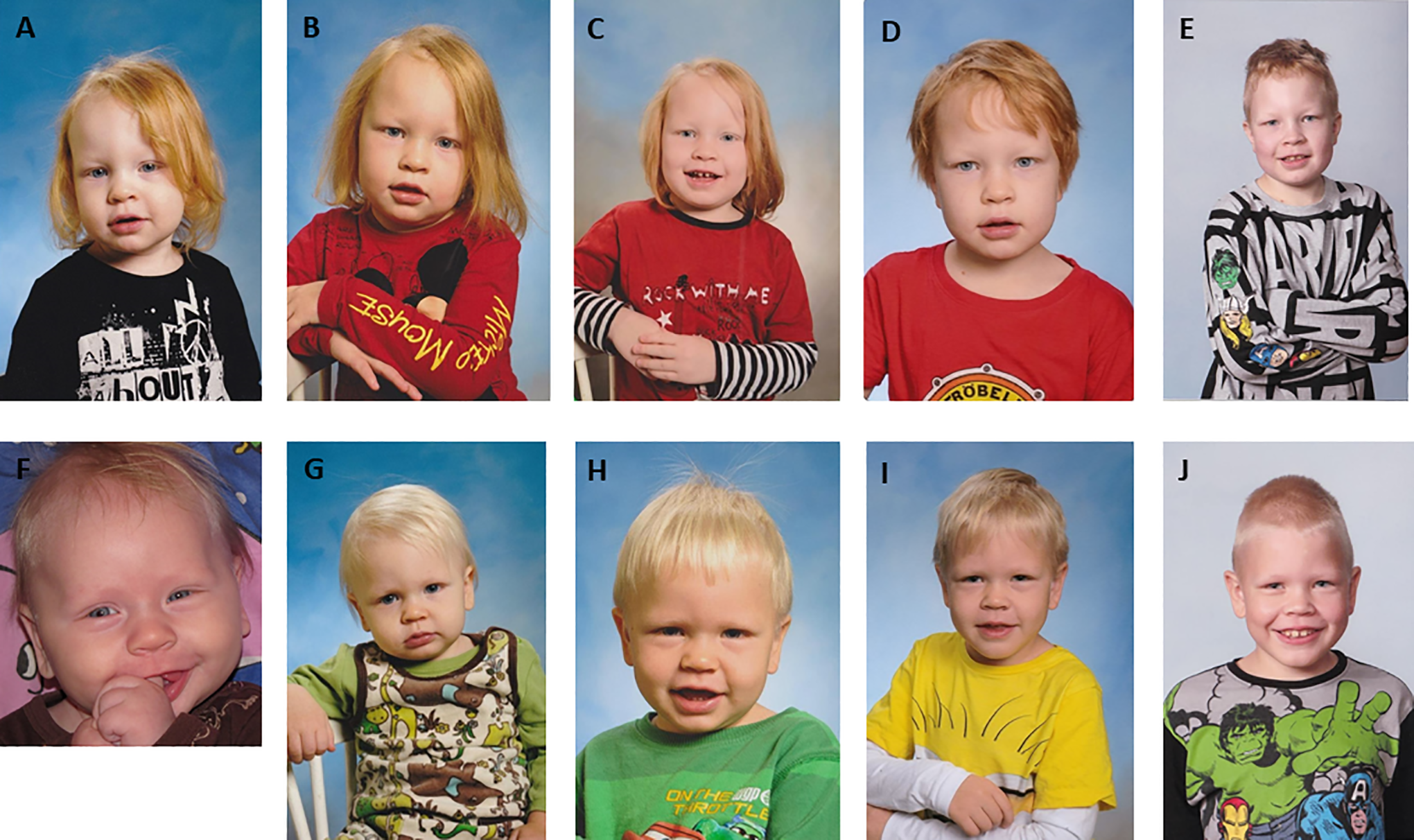
Changes in AGU facial features in 2 brothers over time. (A-E) Patient 1 at 2 years (A), 3 years (B), 4 years (C), 6 years (D), and 10 years (E). (F-J) Patient 2 at 4 months (F), 1 year (G), 2 years (H), 5 years (I), and 9 years (J).

Changes in AGU facial features over time showing normal features in newborns, which become coarser with increasing age. (A-C) Patient 1 at 2 weeks (A), 2 years (B), and 4 years (C). (D-F) Patient 2 at 9 months (D), 5 years (E), and 8 years (F). (G-I) Patient 3 at 5 years (G), 7 years (H), and 12 years (I).

Changes in AGU facial features over time, showing puffy eyelids and shortened nose. Obesity is also common. (A-F) Patient 1 at 2 years (A), 4 years (B), 5 years (C), 9 years (D), 14 years (E), and 17 years (F). (G-L) Patient 2 at 3 years (G), 4 years (H), 5 years (I), 8 years (J), 10 years (K), and 13 years (L).
Developmental characteristics of AGU at an early stage (birth to 5 years) include delays in speech as well as gross and fine motor skills. Children may also display hyperactivity along with intellectual disability that worsens over time. Some children may experience the loss of motor skills previously acquired in early life. The following physical characteristics may be observed in early-stage AGU: early growth spurt, inguinal and abdominal hernias, macrocephaly, clumsiness, and recurring upper respiratory tract infections necessitating tonsillectomy early in life as well as recurrent ear infections requiring multiple placements of tympanostomy tubes. The characteristic facial features of AGU tend to become more noticeable over time, similar to other lysosomal storage disorders. Cognitive delays become more apparent in school-aged children. Adolescent AGU patients may also experience behavioral or mental health issues, and adults have increased risk of seizures. 44 -46 Dental problems may also appear in adolescence and adulthood. 56
Brain MRI may also prove useful in helping to confirm a diagnosis of AGU. 42 Previous studies analyzing the MRI findings in AGU patients have reported the presence of poor myelination and differentiation of cortical gray and white matter, decreased T2 signal intensity in the pulvinar nuclei, increased T2 signal intensity in the periventricular white matter, dilated perivascular spaces, arachnoid and pineal cysts, 43 as well as J-shaped sella turcica and Chiari type 1 malformation. 18
Therapies
Current
No curative therapies currently exist for AGU. In the absence of a curative treatment for AGU, current therapies focus on symptom reduction, which can include medical management of attention-deficit hyperactivity disorder (ADHD), seizures, sleep disorders, and arthritis. Supplemental treatments including speech therapy, occupational therapy, physical therapy, behavioral therapy, and development of individualized educational goals have also been employed to improve adaptive skills and patient independence. 1
Future/Horizon
Potential disease modifying therapies are currently under development. Preclinical studies of enzyme replacement therapy for AGU have shown that transfer of recombinant human aspartylglucosaminidase cleared intracellular aspartylglucosamine, preventing its accumulation in AGU fibroblasts and lymphocytes. 57,58 Intraperitoneal delivery in 1-week-old AGA-null mice reduced aspartylglucosamine levels in the brain by up to 41%. 59 However, no studies in humans have been initiated to date. Despite the preclinical murine data, a potential limitation to enzyme replacement therapy may include inefficient transit across the blood-brain barrier, 59 limiting its neurologic impact. In addition, there may be possible immunologic responses, particularly in the setting of increased doses. 60
Chaperone therapy has also been investigated as a potential therapeutic strategy to facilitate the correct folding of aspartylglucosaminidase mutants given that most, including the AGUFin major variant, cause misfolding of the protein, interfering with protein processing required for activity. 23 In AGU patient fibroblasts, treatment with betaine, a small molecule osmolyte chaperone, increased aspartylglucosaminidase processing as well as enzyme activity. 23 A phase II study is currently underway in Finland to assess the efficacy and safety of betaine in children and adolescents (<18 years) with AGU, preferably with the AGUFin major variant. 61
Although hematopoietic stem cell transplantation (HSCT) has been performed in a small number of AGU patients, the benefit of transplantation is unclear. Studies have shown that it was not successful in improving general health and intellectual disability. 62,63 In 2 siblings with AGU who underwent allogeneic stem cell transplantation from unrelated donors at 10.5 years and 5.8 years, respectively, the absence of psychomotor decline was observed with stable aspartylglucosaminidase expression at 5 years’ follow-up. 64 In addition, improvements in Tau protein, a neuronal and axonal degeneration marker, and white matter composition by MRI was also detected. 64 These observations suggest the potential of HSCT to alter the natural history of AGU. Although donor cells have the potential to migrate to the brain after HSCT, there are disadvantages to HSCT that include side effects from conditioning, risk of graft versus host disease, the potential of graft failure, and insufficient engraftment to some affected tissues, such as bone and heart. 60
As a monogenic disease due to deficiency of a soluble, secreted enzyme, AGU is particularly amenable to viral-mediated gene therapy approaches to restore aspartylglucosaminidase activity, an approach that has been refined over the last decade. 65 In one of the first preclinical studies of gene therapy for AGU, intrastriatal injection of AGA-null mice with an adenovirus-expressing AGA driven by neuron-specific enolase (NSE), astrocyte-specific glial fibrillary acidic protein (GFAP), and the endogenous AGA promoter reduced lysosomal storage as early as 4 weeks post treatment. 66 Similarly, adenovirus-mediated aspartylglucosaminidase expression driven by the Rous sarcoma virus long terminal repeat (LTR) promoter was detected in the liver of AGA-null mice following injection into the tail vein. Intraventricular injection of the same AGA-expressing adenovirus resulted in aspartylglucosaminidase expression in the ependymal cells and neighboring neurons accompanied by correction of the lysosomal storage material defect in both infected brain tissue and neighboring neurons, likely through cross-correction via the mannose-6-phosphate pathway. 67 These pivotal studies demonstrated successful rescue of the biochemical phenotype using a viral-mediated gene therapy approach; however, there were still significant risks associated with adenovirus vectors in this application, especially immunogenicity of the vector itself. Adeno-associated virus (AAV) has proven to be a safer viral vector, and various serotypes have been developed with specific tissue tropism profiles. 68
Preliminary studies evaluating intravenous or intrathecal administration of low and high doses of AAV serotype 9 (AAV9)-harboring AGA in early symptomatic AGA-null mice resulted in complete or near-complete elimination of toxic substrate in central and peripheral tissues and body fluids in a dose-dependent manner with concomitant increased aspartylglucosaminidase activity. 69 Increased mobility and distance traveled were also noted on open-field testing of the AGU-null mice in a dose-dependent manner. Preservation of cerebellar Purkinje cells, which are lost in high levels in AGA-null animals, 70 and reduced gliosis were also detected. 69 This gene therapy approach was well tolerated even at supraphysiological expression levels of the aspartylglucosaminidase enzyme. Advantages to gene therapy include the wide tissue tropism of some viral vectors as well as the potential for a single curative treatment. 71 In addition, preclinical studies have shown that viral vectors, such as AAVs, can cross the blood-brain barrier for systemic transgene expression following intrathecal injections, which allows for smaller dose applications. 72 -75
As potential disease-modifying therapies progress in clinical development for AGU, a validated biomarker may enable a shorter clinical study as there is a lack of disease-specific outcome measures in AGU. An established, validated biomarker may be further utilized as a surrogate biomarker to serve as a “direct measure of how a patient feels, functions or survives.” 76 Levels of aspartylglucosaminidase enzyme activity as well as its substrate, aspartylglucosamine, may potentially serve as surrogate biomarkers in AGU. 77
Challenges
Rarity—Epidemiology
AGU is a rare disorder that affects males and females in equal numbers. AGU is a pan-ethnic disease affecting people from around the world. The majority of reported cases are of Finnish descent, where the prevalence was 160 cases in 5.3 million persons in 2014. 1
Delayed Diagnosis
Although the gene responsible for AGU was identified almost 30 years ago, in non-Finnish populations, AGU diagnoses are often delayed (Table 1). Even in Finland where the carrier frequency is well recognized, AGU is typically not diagnosed until 5 years of age or later. In addition, AGU may be underdiagnosed or misdiagnosed. 32 As previously mentioned, nonspecific findings of developmental delay, including delayed speech or clumsy walking, in early childhood may be an early presenting sign of AGU, especially in the context of additional systemic clinical symptoms (Table 2).
As described previously, additional sporadic variants have been reported in families of diverse ethnic backgrounds in unrelated patients (Table 1). 4 Some patients with novel AGA variants present with classical AGU features, whereas others may present with unique features, especially if AGU is inherited with other autosomal recessive conditions in families with a history of consanguinity. As shown in Table 1, many AGU patients are diagnosed beyond early childhood, likely due to the rarity and geographic dispersion of the variants, which may result in delayed or underdiagnosis.
Comorbidities and Burden
Because of the progressive nature of AGU, the presence of comorbidities, including seizures and arthritis, increases over time, thus escalating the burden of the disease for AGU patients. With age, AGU patients also experience worsening cognitive impairment, 33 loss in independence and socialization, and sleep disturbances, 36 along with increased behavioral problems. 35 This can place greater burden on not only the patients but also their caregivers, impacting the work day and productivity of caregivers. In addition, carriers of AGA variants may be more likely to suffer from chronic arthritis. 48
No study has analyzed the burden of AGU; however, similar analyses in autism and ADHD may provide insights into the potential impact of this disease on the patients, families, and society as a whole. Analysis of the economic burden of autism in the United States, including health care, school, therapy, family services, and caregiving costs, showed that it was associated with $3020 higher health care costs per year and $14 061 higher non–health care costs per year compared to children without autism. 78 The severity of disability in AGU may also impact caregiver quality of life (QoL), given that up to two-thirds of parents of young adults with autism spectrum disorder reported that the disorder had a moderate to high impact on their QoL, particularly their emotional QoL. 79 Furthermore, greater reductions in caregiver QoL were related with reduced levels of adaptive skills (eg, receptive, expressive, and written language skills, daily living skills, and socialization), increasing symptom severity, and the presence of challenging behaviors in their children. 79 Conversely, parents of children with improvements in irritability, lethargy, stereotypy, and hyperactivity reported a decreased impact on QoL. 79
Early Diagnosis
Clinical Impact—Sense of Urgency
Although there is no curative treatment for AGU at present, early diagnosis of AGU has potential to provide benefit both to patients and their caregivers in terms of anticipatory guidance, setting expectations, early interventions, symptom management, and information on end-of-life decisions. For example, early initiation of interventions, including speech and occupational therapy, may improve social skills and patient independence. 1 Development of patient-specific learning goals in younger children may reduce caregiver burden. Patients and caregivers may also benefit from peer support groups with an early diagnosis.
Conclusion
AGU is a severe and progressive autosomal recessive lysosomal storage disorder. Because of low awareness and high prevalence of autism and ADHD, AGU may be underdiagnosed or patients may initially be diagnosed with autism spectrum disorder before receiving the diagnosis of AGU, especially in non-Finnish populations. The natural history of AGU is characterized by progressive intellectual disability, connective tissue abnormalities along with arthritis, seizures, frequent infections, and behavioral changes, ultimately leading to premature death. Preclinical studies are paving the way for promising new disease-modifying treatments with the potential to change the natural course of AGU. The increased utilization of early genetic testing for AGU also holds promise for early diagnosis, which will be imperative in obtaining maximal clinical benefits from future disease-modifying therapies.
Footnotes
Acknowledgments
The authors thank the patients and their families for their participation. Medical writing assistance was provided by Marjet Heitzer, PhD, of 360 Medical Writing and supported by Neurogene Inc.
Author Contributions
KG contributed to the developing, drafting, and revising the manuscript. CF contributed to manuscript conception and editing. ML contributed to providing clinical information, drafting and editing the manuscript. TCL contributed to the developing, drafting, and revising the manuscript.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: KG reports consultancy fees and research support from Neurogene. CF is an employee of Neurogene. ML and TCL report consultancy fees from Neurogene.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Neurogene provided financial support for the development of this review.
Ethical Approval
Written informed consent was obtained from the patients and patients’ parents for publication of the images in Figures 1
–![]() . A copy of written consent is available for review by the Editor-In-Chief of this journal.
. A copy of written consent is available for review by the Editor-In-Chief of this journal.