
Abstract
Hepatic encephalopathy (HE) is a frequent and life-threatening neurological complication of acute liver failure (ALF) encountered in the intensive care unit and remains a major determinant of short-term mortality and long-term neurological outcomes. Increasing evidence indicates that HE in ALF is primarily driven by hyperammonemia, with synergistic contributions from systemic inflammation, cerebral hemodynamic dysregulation, metabolic failure, and osmotic imbalance. These interacting mechanisms promote astrocytic swelling, disruption of the blood–brain barrier, cerebral edema, and intracranial hypertension. In patients with ALF, early recognition of cerebral involvement and systematic exclusion of alternative causes of altered mental status are essential. Given the dynamic and heterogeneous neurological manifestations of HE, reliance on isolated clinical, biochemical, or radiological parameters is insufficient. Multimodal neuromonitoring—integrating neurological examination, ammonia kinetics, cerebral hemodynamic assessment, and neuroimaging—allows more accurate assessment of cerebral injury and supports timely, targeted intervention. This review summarizes current evidence on the pathophysiology of HE in ALF with a focus on mechanisms relevant to intensive care practice. We highlight evidence-based strategies for cerebral protection, including early and sustained control of hyperammonemia with continuous renal replacement therapy, optimization of cerebral perfusion and osmotic balance, selective use of plasma exchange, and structured neurocritical care. An integrated management framework is proposed to guide prognostication and inform timely decisions regarding advanced liver support and liver transplantation in the ICU setting.
Introduction
Hepatic encephalopathy (HE) is a major neurological complication of acute liver failure (ALF) with a broad clinical spectrum and significant impact on patient outcomes.1,2 Cumulative organ failure, especially neurologic and respiratory failure, significantly impacts waitlist and post-LT mortality in patients with ALF and may inform risk-prioritized allocation of organs. 3 Both adult and pediatric survivors, including those who successfully undergo liver transplantation, may experience persistent neurocognitive impairment, behavioral abnormalities, and reduced quality of life. These observations suggest that ALF-related brain injury is not fully reversible, underscoring cerebral protection as a central therapeutic goal in the management of ALF. Despite improvements in organ supportive care and liver transplantation in ALF, severe HE and cerebral complications remain major contributors to reduced transplant-free survival and poorer post-transplant outcomes.4,5 Etiology-specific neurological outcomes have been observed after LT for ALF, with acetaminophen-related ALF showing a higher risk of post-transplant brain death, and adenovirus-associated PALF during the 2022 outbreak presenting with more severe HE and clinical instability despite similar survival.6,7
Early identification of cerebral edema and intracranial hypertension is critical for reducing morbidity and mortality in acute liver failure. Given the heterogeneous neurological manifestations of hepatic encephalopathy in acute liver failure, reliance on a single clinical or radiological parameter may be misleading. Emerging evidence suggests that multimodal neuromonitoring can improve diagnostic accuracy. 8 Such an approach enables more timely and targeted interventions and may ultimately improve neurological outcomes in this high-risk population. When evaluating hepatic encephalopathy, it is essential to systematically exclude alternative causes of altered mental status. These include, but are not limited to, respiratory failure, electrolyte disturbances, seizures or status epilepticus, renal failure, stroke, hypoglycemia, central nervous system infections, metabolic acidosis, and drug intoxication. 9
Recent studies increasingly demonstrate that cerebral dysfunction in ALF is not attributable to a single mechanism but rather results from the convergence of multiple pathophysiological processes, including hyperammonemia, systemic inflammation, dysregulation of cerebral blood flow, impaired energy metabolism, osmotic disturbances, and upregulation of aquaporin-4 (AQP4) expression.10-13 A systematic understanding of these mechanisms, together with the development of effective monitoring and intervention strategies, is critical for optimizing cerebral protection in ALF.
Mechanisms of Hepatic Encephalopathy in Acute Liver Failure
HE in ALF is characterized by rapid neurological deterioration and is predominantly driven by hyperammonemia. Loss of hepatic urea synthesis transforms the liver from an ammonia-clearing organ into a net producer of ammonia, resulting in markedly elevated circulating levels. In this setting, hepatic venous ammonia concentrations may exceed arterial levels, reflecting impaired detoxification and increased systemic ammonia burden. Elevated ammonia is closely associated with the development of cerebral edema and the severity of HE.14,15
Ammonia exerts its neurotoxic effects primarily on astrocytes, where its metabolism to glutamine increases intracellular osmotic pressure, leading to cellular swelling and dysfunction. This process is characterized by the presence of Alzheimer type II astrocytes and represents a key cellular mechanism linking hyperammonemia to cerebral edema and intracranial hypertension.16-20
Beyond ammonia, systemic inflammation plays a critical synergistic role in ALF-related brain injury. Inflammatory mediators alter cerebral endothelial function, vascular tone, and astrocyte activity, thereby increasing blood–brain barrier permeability to neurotoxins. Pronounced innate immune activation and elevated cytokine levels have been observed in ALF and are closely associated with multiorgan failure and neurological deterioration. 11 Disruption of the blood–brain barrier facilitates the entry of neurotoxic substances into the brain parenchyma, further exacerbating astrocyte injury. Microglia and astrocytes contribute to this process through cell-specific inflammatory responses, while cytokine and chemokine signaling orchestrates neuroimmune activation. In addition, the choroid plexus and meninges have emerged as important interfaces linking circulating ammonia and systemic inflammation to central nervous system injury, underscoring the pivotal role of neuroinflammation in HE. 21
Cerebrospinal fluid metabolomic analyses have identified alterations in 73 metabolites among patients with HE, including amino acids, acylcarnitines, bile acids, and nucleosides. A hallmark feature is disrupted energy metabolism, characterized by accumulation of acetylated compounds, likely related to tricarboxylic acid cycle dysfunction. 14 Experimental evidence further supports a central metabolic component in HE pathogenesis. In a rat model of ALF induced by subtotal hepatectomy, cerebrospinal fluid levels of glutamine and albumin showed strong correlations with neurological severity, outperforming other amino acids. 15 In addition, cerebral lactate accumulation suggests a state of “cytopathic hypoxia,” wherein mitochondrial dysfunction leads to energy failure despite preserved oxygen delivery.
At the cellular level, osmotic dysregulation is mediated in part by aquaporin-4 (AQP4), which is predominantly expressed at astrocytic end-feet and plays a central role in cerebral water homeostasis. In ALF, upregulation of AQP4 facilitates excessive water movement along osmotic gradients, thereby amplifying astrocytic swelling and cerebral edema. 16 Animal studies demonstrate that AQP4 knockout mice exhibit significant protection in liver failure models, with preserved brain water content and attenuated neurological deficits compared with wild-type controls, implicating AQP4 as a key mediator of astrocytic swelling 17 (Figure 1).
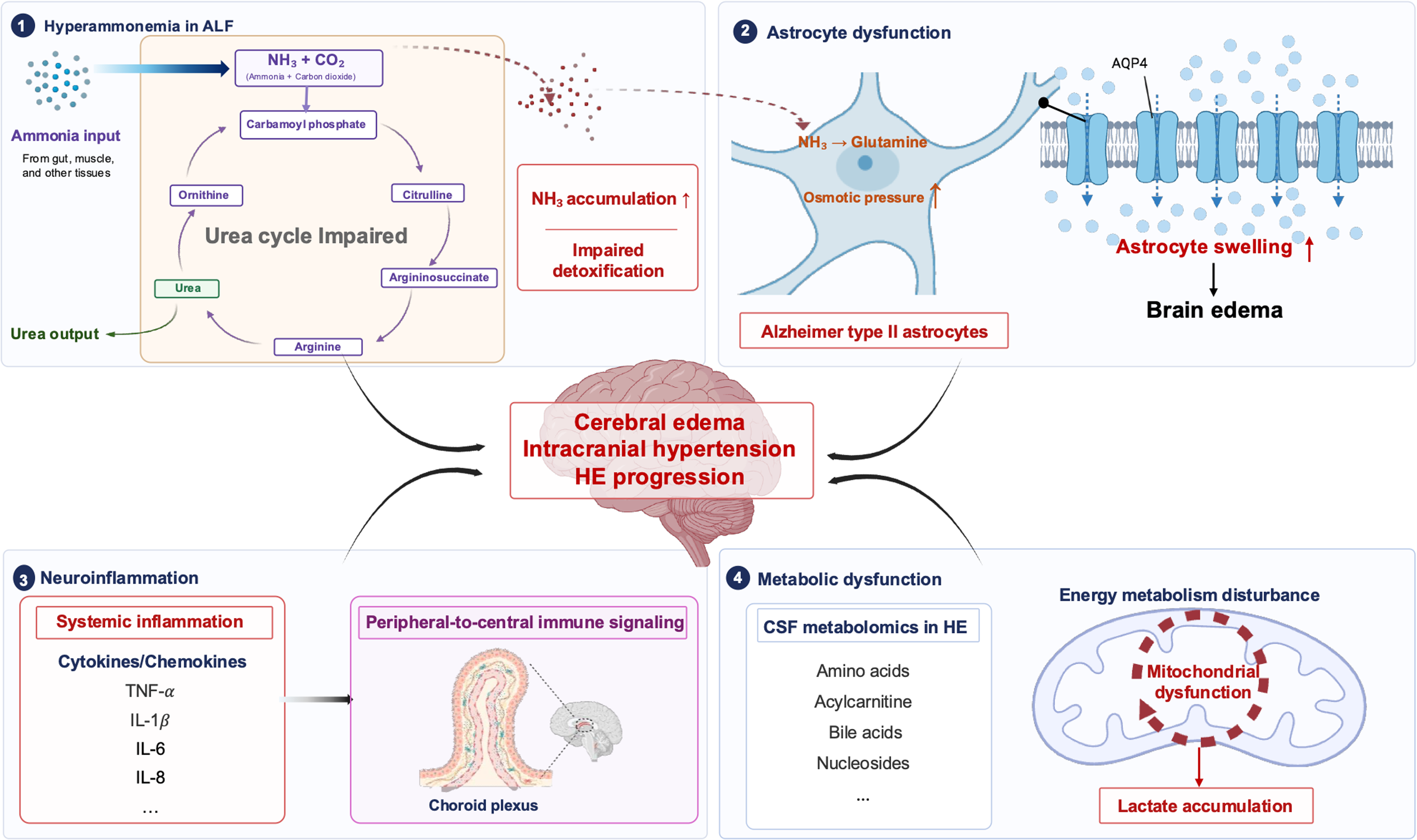
Pathophysiological mechanisms of hepatic encephalopathy in ALF.
Assessment of Cerebral Function in Acute Liver Failure
History and Physical
Hepatic encephalopathy ranges from subtle cognitive impairment and sleep disturbance to severe neuromuscular dysfunction. Early signs include reduced alertness and attention deficits, while progression is marked by asterixis, hyperreflexia, myoclonus, and ultimately coma. 18
In ALF, neurological examination is critical for risk stratification. Although absent pupillary reflexes indicate poor prognosis, neurological recovery remains possible after transplantation. 19
Imaging Evaluation
Radiologic imaging, such as CT or MRI, may be used to rule out alternative causes of encephalopathy, including intracranial lesions, masses, or hemorrhage. In a large multicenter cohort from the U.S. Acute Liver Failure Study Group, cerebral edema was identified on brain CT scan in 43.2% of ALF patients who underwent neuroimaging, and 4.5% subsequently developed tonsillar herniation. The presence of cerebral edema on CT was independently associated with worse 21-day transplant-free survival, although it did not preclude liver transplantation. 20 Typical MRI findings in HE include bilateral basal ganglia hyperintensities related to manganese deposition in chronic liver disease, as well as diffuse hemispheric white matter abnormalities on T2-weighted, fluid-attenuated inversion recovery, or diffusion-weighted imaging, frequently involving the corticospinal tracts. 21 In ALF-HE, reduced apparent diffusion coefficient values on diffusion-weighted imaging may be observed, consistent with cytotoxic edema.
Hyperammonemia
Clinically, HE in ALF should be viewed as a dynamic continuum, in which functional cerebral impairment may rapidly progress to structural brain injury, intracranial hypertension, and cerebral herniation. In a prospective study, Kumar et al demonstrated that persistent hyperammonemia (≥122 μmol/L for three consecutive days) was strongly associated with cerebral edema, seizures, infections, and a markedly increased risk of mortality compared with isolated baseline hyperammonemia (odds ratio [OR] 10.7 vs 2.4). 10 Notably, even sustained moderate hyperammonemia (≥85 μmol/L) significantly increased complication rates. In a multicenter Chinese pediatric acute liver failure (PALF) cohort, admission ammonia level emerged as the sole independent predictor of mortality, with incremental increases in ammonia conferring a stepwise rise in death risk. 22 Among patients with PALF, the magnitude of ammonia reduction within 48 h was strongly associated with survival; each 10% decrease in ammonia was associated with an approximately 50% increase in survival probability. 12 These studies underscore the importance of serial ammonia monitoring for neurological risk stratification and therapeutic decision-making in ALF. In our clinical experience, arterial ammonia measurements appear to be more accurate and clinically informative than venous levels. It should be noted that these data are derived from PALF cohorts and may not be directly generalizable to adult populations, warranting cautious interpretation.
Systemic Inflammation
Markers of systemic inflammation provide complementary information for the assessment of cerebral involvement in ALF. A multicenter study in children with ALF demonstrated that serum S100β was significantly associated with the presence of hepatic encephalopathy, with inflammatory activation, particularly elevated interleukin-6, also showing a significant association. These findings suggest that circulating neurological and inflammatory biomarkers may aid in the assessment of cerebral involvement in pediatric acute liver failure. 13 Circulating inflammatory mediators, including tumor necrosis factor-α (TNF-α) and interleukins such as IL-1β and IL-6, have been shown to correlate with neurological deterioration and adverse clinical outcomes in ALF. 23 In addition, interventional and experimental studies demonstrate that therapies and conditions associated with attenuation of systemic inflammatory activity are accompanied by reduced central nervous system complications, indirectly supporting the clinical relevance of inflammatory burden as a measurable risk marker.
Dysregulation of Cerebral Blood Flow and Energy Metabolism
Under physiological conditions, cerebral perfusion is maintained by intact autoregulation and the neurovascular unit. In ALF, however, circulatory failure, infection, and renal dysfunction frequently coexist, and cerebral autoregulation is profoundly impaired during HE. A meta-analysis suggested that hepatic encephalopathy is associated with impaired cerebral energy metabolism, characterized by reduced cerebral metabolic rates of oxygen and glucose and decreased cerebral blood flow.24,25 Cerebral hemodynamic disturbances represent an important but often underrecognized component of HE in ALF. A prospective study using transcranial Doppler demonstrated that cerebral autoregulation (CA) was markedly impaired in ALF prior to LT. Importantly, CA gradually recovered after transplantation, with re-establishment observed in most patients within 48–72 h post-transplant, paralleling neurological stabilization. 26 In a single-center retrospective cohort study of 106 patients with ALF listed for emergency liver transplantation, transcranial Doppler (TCD) monitoring was performed in 44% of patients during intensive care unit stay. Abnormal TCD findings, defined as a pulsatility index (PI) > 1.2, were observed in more than half of the examined patients and were associated with more severe hepatic and extrahepatic organ failure. 27 TCD may be a useful non-invasive tool for ICH detection and management, then guide sedation withdrawal.
Among patients who died prior to liver transplantation (LT), intracranial pressure (ICP) monitoring was more frequently required. In addition, ICP-directed therapies—including mannitol, barbiturates, therapeutic hypothermia, and sedatives—were used significantly more often than in survivors. These patients also experienced higher rates of intensive care unit complications. 28 Compared with one-year survivors after liver transplantation, non-survivors were more likely to require pretransplant ICP monitoring and ICP-directed therapies.
In a retrospective cohort of pediatric acute liver failure from Northeast China, early lactate–albumin ratio (LAR), reflecting the combined effects of tissue hypoperfusion and impaired hepatic synthetic function, was significantly associated with survival with the native liver. Notably, the prognostic impact of LAR was more pronounced in patients with higher-grade hepatic encephalopathy, with a significant interaction observed between LAR and HE severity. 29 Disturbances in cerebral blood flow and energy metabolism represent key components of neurological dysfunction in ALF. Bedside assessment using non-invasive cerebral hemodynamic monitoring and simple metabolic biomarkers may complement clinical examination and biochemical indices for neurological risk stratification and prognostic assessment (Figure 2).
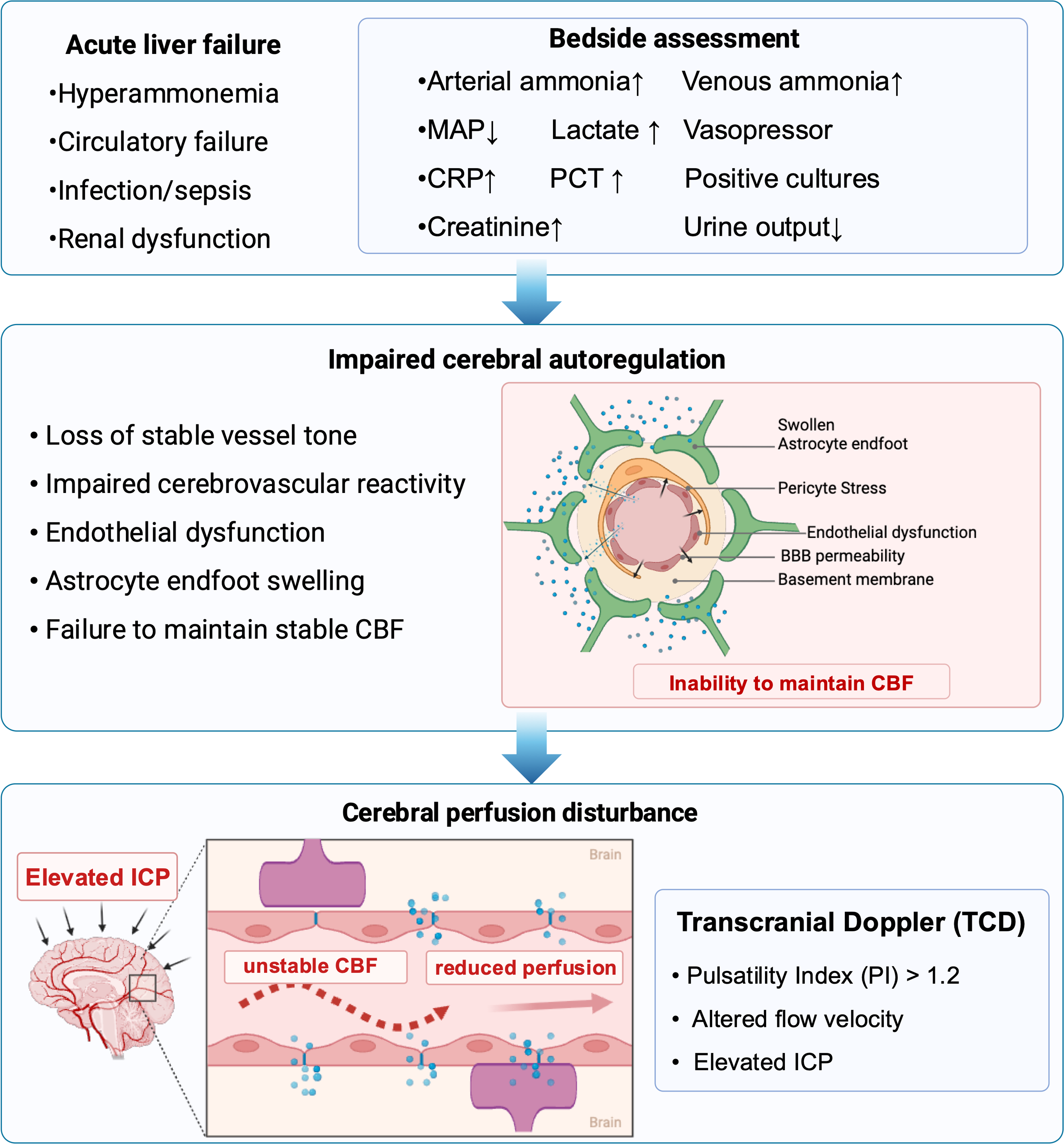
Dysregulation of cerebral blood flow and cerebral autoregulation in ALF.
Osmotic Disturbances
A retrospective study in patients with acute and acute-on-chronic liver failure suggested that elevated serum osmolality is independently associated with greater severity of hepatic encephalopathy. Increased osmolality was also linked to altered cerebrospinal fluid density, supporting a potential role for osmotic disturbances in blood–brain barrier dysfunction and cerebral toxin exposure. 30
More importantly, acute declines in osmolality are strongly linked to cerebral swelling and neurological deterioration. In one study with a median baseline osmolality of 310 mOsm/kg, an acute decrease of 9 mOsm/kg was associated with radiographic evidence of increased brain volume in 68% of patients. Changes in osmolality correlated independently with cerebrospinal fluid volume (r = 0.70) and GCS deterioration. 31
Strategies for Cerebral Protection in Acute Liver Failure
Control of Hyperammonemia
Effective control of hyperammonemia in ALF centers on continuous renal replacement therapy (CRRT), supplemented by plasma exchange when appropriate. Traditional gut-directed therapies have limited evidence in ALF. Lactulose remains first-line therapy for HE in chronic liver disease but is only appropriate in ALF patients with mild encephalopathy (grade I–II) who can safely protect their airway, with caution regarding pre-transplant bowel distension. 32
Rifaximin combined with lactulose is superior to lactulose alone in chronic liver disease but lacks direct evidence in ALF, and its use is largely extrapolative. L-ornithine-L-aspartate (LOLA), despite theoretical benefits, failed to demonstrate superiority over standard therapy or placebo in randomized trials and is not recommended in ALF. 33 Similarly, recent trials of ornithine phenylacetate have not confirmed clinical efficacy.
General Neurocritical Care Measures
All patients with ALF should receive standardized neuroprotective care, including head-of-bed elevation to 30°, neutral head positioning, avoidance of hypoxia, hypotension, hypoglycemia, and PaCO₂ disturbances, maintenance of serum sodium at 140–145 mmol/L, hemodynamic stability, and aggressive infection control.
Sedation and Airway Management
Patients with grade III–IV HE or GCS < 8 should undergo early intubation and sedation to protect the airway and prevent agitation-induced increases in intracranial pressure. Short-acting sedatives such as propofol or dexmedetomidine are preferred due to rapid offset and minimal encephalopathy exacerbation. 34
Temperature Management
The primary goal is strict fever avoidance, maintaining core temperature at 35–36°C. Induced hypothermia (32-34 °C) should be reserved for refractory intracranial hypertension and not used prophylactically, given the lack of survival benefit and increased risk of infection, arrhythmia, bleeding, and impaired liver regeneration.35,36
Optimization of Hemodynamics and Cerebral Perfusion
ALF is characterized by a hyperdynamic, vasodilatory circulatory state resembling septic shock. In patients with intracranial hypertension, mean arterial pressure should be maintained at 70–80 mm Hg to preserve cerebral perfusion pressure. Norepinephrine is the first-line vasopressor, with vasopressin as adjunctive therapy when needed.37,38 In the ICU management of ALF, bedside noninvasive monitoring—such as optic nerve sheath diameter, transcranial Doppler, and continuous EEG-is increasingly replacing repeated CT-based patient transfers and has become the primary approach for continuous neurological monitoring.39,40 During the waiting period for liver transplantation, precise guidance of cerebral perfusion pressure (CPP) management is required, with a target CPP > 50–60 mm Hg. 41 A TCDpulsatility index (PI) > 1.2 is significantly associated with mortality or the need for liver transplantation. 42 An optic nerve sheath diameter (ONSD) > 6 mm in adults and >4.55 mm in children suggests elevated ICP. 43
Osmotic Therapy
Hypertonic saline is preferred over mannitol for both prevention and treatment of intracranial hypertension, with a target serum sodium of 145–155 mmol/L. Compared with mannitol, hypertonic saline (continuous infusion of 3% hypertonic saline) offers a lower risk of rebound cerebral edema and acute kidney injury.44,45 When mannitol is used, serum osmolality should be maintained at <320 mOsm/L. The rate of correction should not exceed 6–8 mmol/L per 24 h, and the upper limit of serum sodium should not exceed 160 mmol/L. 46
Plasma Exchange
High-volume plasma exchange has been shown in multicenter randomized trials to improve transplant-free survival and reduce systemic inflammation in ALF. 11 Current guidelines suggest considering plasma exchange in severe ALF with hyperammonemia when resources permit, although optimal dosing and intensity remain uncertain. 45
Continuous Renal Replacement Therapy
CRRT is a cornerstone of cerebral protection in ALF, with indications extending beyond acute kidney injury to isolated hyperammonemia. Early initiation, particularly within 4–6 h of ICU admission, significantly reduces extreme hyperammonemia and improves transplant-free survival.38,47
Advanced Liver Support Systems
Bioartificial liver systems can reduce ammonia and cerebral edema but remain investigational, with no proven survival benefit in human ALF. Current guidelines restrict their use to clinical trials.46,48
Corticosteroids
Corticosteroids are not routinely recommended for cerebral protection in ALF and may be harmful in patients with high MELD scores (>40). Potential benefit appears limited to selected populations, including autoimmune hepatitis-related ALF and immune-activated pediatric ALF.49,50
An Evidence-Based Framework for Cerebral Protection in ALF
Building upon the therapeutic strategies outlined above, cerebral protection in acute liver failure should be operationalized as a time-dependent, decision-oriented clinical pathway, rather than a collection of isolated interventions.
Early Phase (ICU Admission to 24 h): Immediate Stabilization and Ammonia Control
We should prioritize rapid identification of patients at high risk of intracranial hypertension and early initiation of targeted therapies. Among all variables, arterial ammonia serves as the central actionable biomarker. Current evidence supports early initiation of CRRT in ALF who develop hyperammonemia (particularly >150 μmol/L) and/or accompanied by grade III–IV encephalopathy or early signs of cerebral edema, even in the absence of acute kidney injury. The optimal timing is within 4–6 h of ICU admission.38,47 Standard effluent flow rates are 35–45 mL/kg/h, with high-volume therapy up to 90 mL/kg/h when necessary; therapeutic efficacy appears to be more closely related to cumulative treatment duration than to intensity alone. The target is to reduce serum ammonia to <150 μmol/L, ideally <100 μmol/L.32,38
At this stage, therapeutic decisions should not be made in isolation. The presence of systemic inflammation, hemodynamic instability, or worsening encephalopathy should trigger early combination strategies, such as the addition of plasma exchange to CRRT. 51 Similarly, rapid neurological deterioration or inability to protect the airway should prompt timely intubation, enabling controlled ventilation and prevention of secondary brain injury.
Close Monitor Phase (Ongoing from Admission): Intgrated Neuroprotective Monitoring and Physiologic Control
Beyond individual interventions, continuous reassessment and integration of physiological signals are essential. Rising ammonia levels, abnormal TCD findings, or clinical progression of encephalopathy should prompt immediate re-evaluation of multiple domains simultaneously—including cerebral perfusion (MAP targets), osmotic therapy (serum sodium targets), and ventilatory control (PaCO₂ targets). The core principle of mean arterial pressure (MAP) management in ALFis an individualized, CPP-guided approach. In the absence of intracranial hypertension, maintaining MAP ≥65 mm Hg is generally sufficient. In patients with established or high risk of intracranial hypertension, the MAP target should be increased to ≥75 mm Hg (or 70-80 mm Hg) to ensure a cerebral perfusion pressure of 60–80 mm Hg. 38 At the same time, end-organ perfusion should be dynamically assessed using multiple parameters, including lactate, urine output, and transcranial Doppler, to avoid indiscriminate escalation of MAP. This coordinated adjustment is critical, as intracranial hypertension in ALF is driven by the interaction of hyperammonemia, inflammation, and cerebral hemodynamic dysregulation, rather than a single pathway.
Neuroimaging and advanced monitoring should be applied selectively and in response to clinical evolution. Rather than routine use, CT imaging should be reserved for acute neurological deterioration or pre-transplant assessment, while invasive ICP monitoring should be limited to highly selected patients with refractory intracranial hypertension in experienced centers. Non-invasive bedside tools, including TCD and ONSD, may provide continuous functional assessment and guide dynamic management.
Reassessment Phase (48-72 h): Dynamic Prognosis and Escalation Decisions
At 48–72 h, trends in arterial ammonia, alpha-fetoprotein, and organ failure scores should guide reassessment of hepatic regeneration potential. A trajectory characterized by persistent hyperammonemia, worsening encephalopathy, and progressive multiorgan failure indicates limited reversibility of cerebral injury. In such cases, prolonged escalation of supportive therapy is unlikely to be beneficial, and expedited decisions regarding liver transplantation or advanced liver support systems should be prioritized.
In this framework, the key principle is integration rather than escalation alone. Effective cerebral protection in ALF depends not only on early initiation of therapies but also on the ability to dynamically adjust them in response to evolving physiological signals, thereby preventing the transition from potentially reversible cerebral dysfunction to irreversible brain injury (Table 1).
Practical Decision Thresholds for Cerebral Protection in Acute Liver Failure.
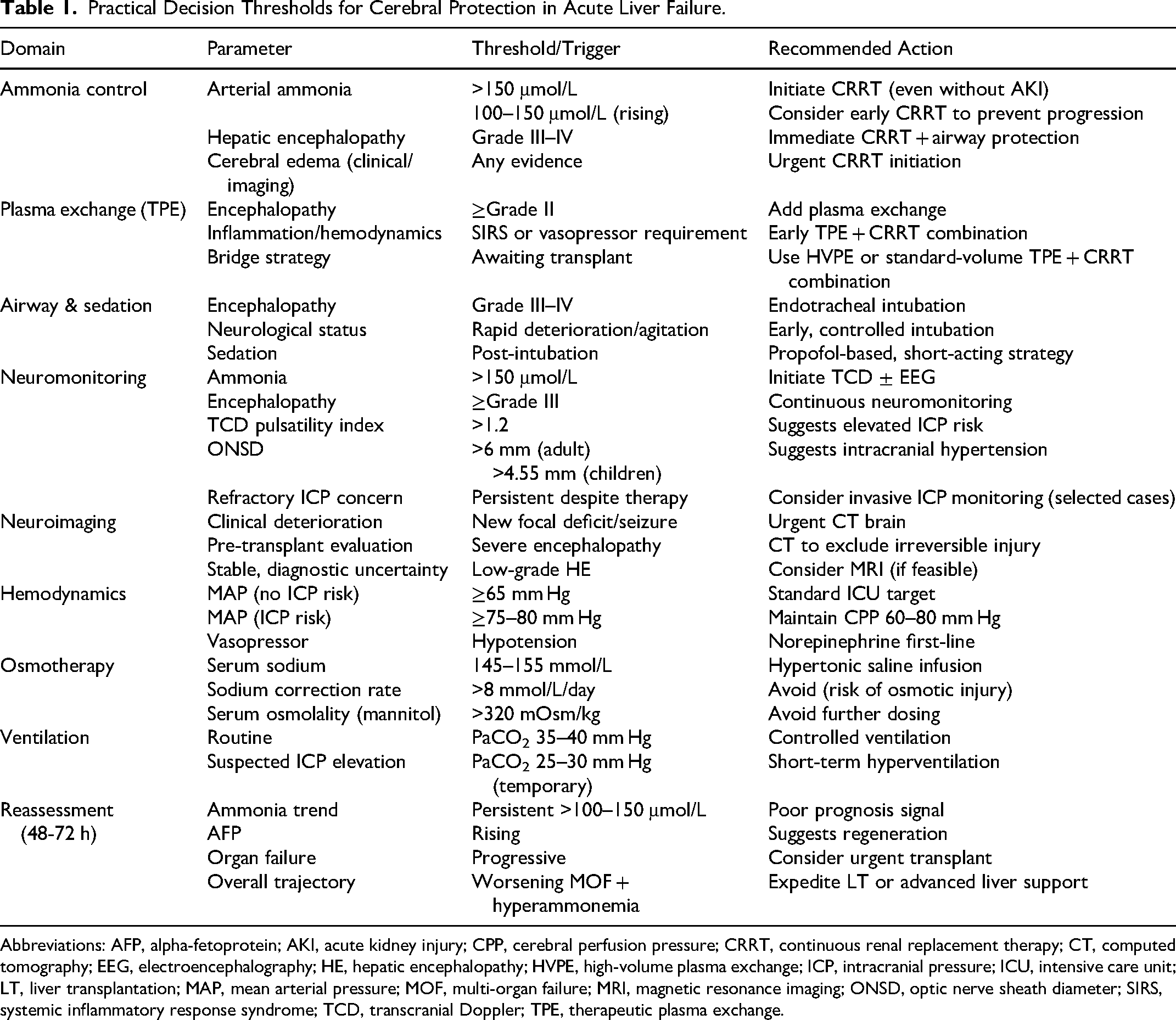
Abbreviations: AFP, alpha-fetoprotein; AKI, acute kidney injury; CPP, cerebral perfusion pressure; CRRT, continuous renal replacement therapy; CT, computed tomography; EEG, electroencephalography; HE, hepatic encephalopathy; HVPE, high-volume plasma exchange; ICP, intracranial pressure; ICU, intensive care unit; LT, liver transplantation; MAP, mean arterial pressure; MOF, multi-organ failure; MRI, magnetic resonance imaging; ONSD, optic nerve sheath diameter; SIRS, systemic inflammatory response syndrome; TCD, transcranial Doppler; TPE, therapeutic plasma exchange.
Conclusion
Hepatic encephalopathy in acute liver failure is a dynamic neurological syndrome resulting from the interplay of hyperammonemia, systemic inflammation, cerebral hemodynamic dysfunction, metabolic failure, and osmotic imbalance. Cerebral injury may progress rapidly and is often incompletely reversible, highlighting cerebral protection as a key therapeutic priority. Early recognition of neurological involvement, exclusion of alternative causes, and multimodal neuromonitoring are essential. Sustained ammonia reduction remains the cornerstone of management. Integrating neurological assessment with biochemical and organ failure can guide timely decisions regarding liver transplantation and advanced liver support to improve survival and neurological outcomes.
Supplemental Material
sj-docx-1-jic-10.1177_08850666261460778 - Supplemental material for Assessment and Protection of Cerebral Function in Patients with Acute Liver Failure
Supplemental material, sj-docx-1-jic-10.1177_08850666261460778 for Assessment and Protection of Cerebral Function in Patients with Acute Liver Failure by Hao-Feng Xiong, Wan-Ting Zhang, Zhi-Jun Zhu and Li-Ying Sun in Journal of Intensive Care Medicine
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (grant number No.82300686).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.