
Abstract
Protein-based biomaterials are widely used in tissue engineering and regenerative medicine; however, many conventional materials suffer from limitations including immunogenicity, low stability, and risks associated with animal-derived sources. In this context, keratin derived from human hair has attracted increasing attention due to its inherent biocompatibility and cysteine-rich structure, which provides reactive thiol (–SH) groups suitable for chemical modification. In the present study, reductively extracted keratin was utilized to preserve thiol functionality, enabling site-selective modification via thiol–maleimide chemistry using an aminoethyl maleimide (AEM) linker. Through this approach, heparin, a clinically established anticoagulant, was immobilized onto keratin films under mild aqueous conditions. The resulting keratin–heparin films exhibited effective suppression of thrombin-induced coagulation, whereas unmodified keratin showed no anticoagulant activity. Furthermore, spectroscopic and colorimetric analyses confirmed successful heparin conjugation and surface presentation, and no measurable heparin release was detected, indicating stable surface immobilization. Overall, these results demonstrate that thiol-specific functionalization provides a selective, stable, and animal-free platform for anticoagulant surface modification, highlighting the potential of keratin-based materials for blood-contacting biomedical applications.
Keywords
Introduction
Tissue engineering, which aims to restore or replace damaged tissues, relies heavily on the development of biomaterials that can provide both structural support and biological signals for cell growth, differentiation, and regeneration.1,2 Among naturally derived polymers, collagen has been extensively studied because of its excellent biocompatibility, biodegradability, and ability to promote cell adhesion.3–5 However, collagen-based materials still face several limitations, including low mechanical and thermal stability, limited controllability of degradation, and batch-to-batch variation.6–10 Furthermore, the animal origin of collagen raises concerns regarding immunogenic responses and possible pathogen transmission.11,12 These drawbacks have motivated researchers to explore safer, sustainable, and animal-free biomaterials that can offer comparable biological performance for clinical use.
Among the promising candidates, keratin has gained increasing attention. Keratin is a fibrous structural protein found in epithelial tissues such as hair, wool, and nails, and it exhibits natural biocompatibility and biodegradability.13–15 In addition, keratin contains specific amino-acid motifs such as RGD and LDV, which support cell adhesion and proliferation.16–18 A distinct advantage of keratin lies in its high cysteine content, forming numerous disulfide bonds (S–S) that contribute to its structural integrity and provide reactive thiol (–SH) groups after reduction.19–21 These thiol groups enable site-selective chemical modification under mild aqueous conditions, allowing keratin to serve as a versatile platform for biofunctionalization. As a result, keratin has been explored as a biomaterial for applications ranging from regenerative scaffolds to surface coatings and drug-delivery systems.
Keratin can be obtained through oxidative or reductive extraction, yielding two different molecular forms—keratose and keratein, respectively. The schematic illustration of these extraction processes is shown in Figure 1. In the oxidative route, disulfide bonds are irreversibly cleaved to form sulfonic acid (–SO3H) groups, producing a water-soluble but chemically inert protein Ref. 22. In contrast, the reductive method cleaves disulfide bonds reversibly, generating thiol-rich keratin that preserves molecular integrity and reactive functionality.23–25 The preservation of free thiol groups provides an effective handle for covalent immobilization of bioactive molecules under biocompatible conditions. Comparison of keratin molecules and extraction mechanisms. Schematic illustration of oxidative extraction (irreversible cleavage of disulfide bonds forming keratose) and reductive extraction (reversible cleavage generating keratein with reactive thiol groups).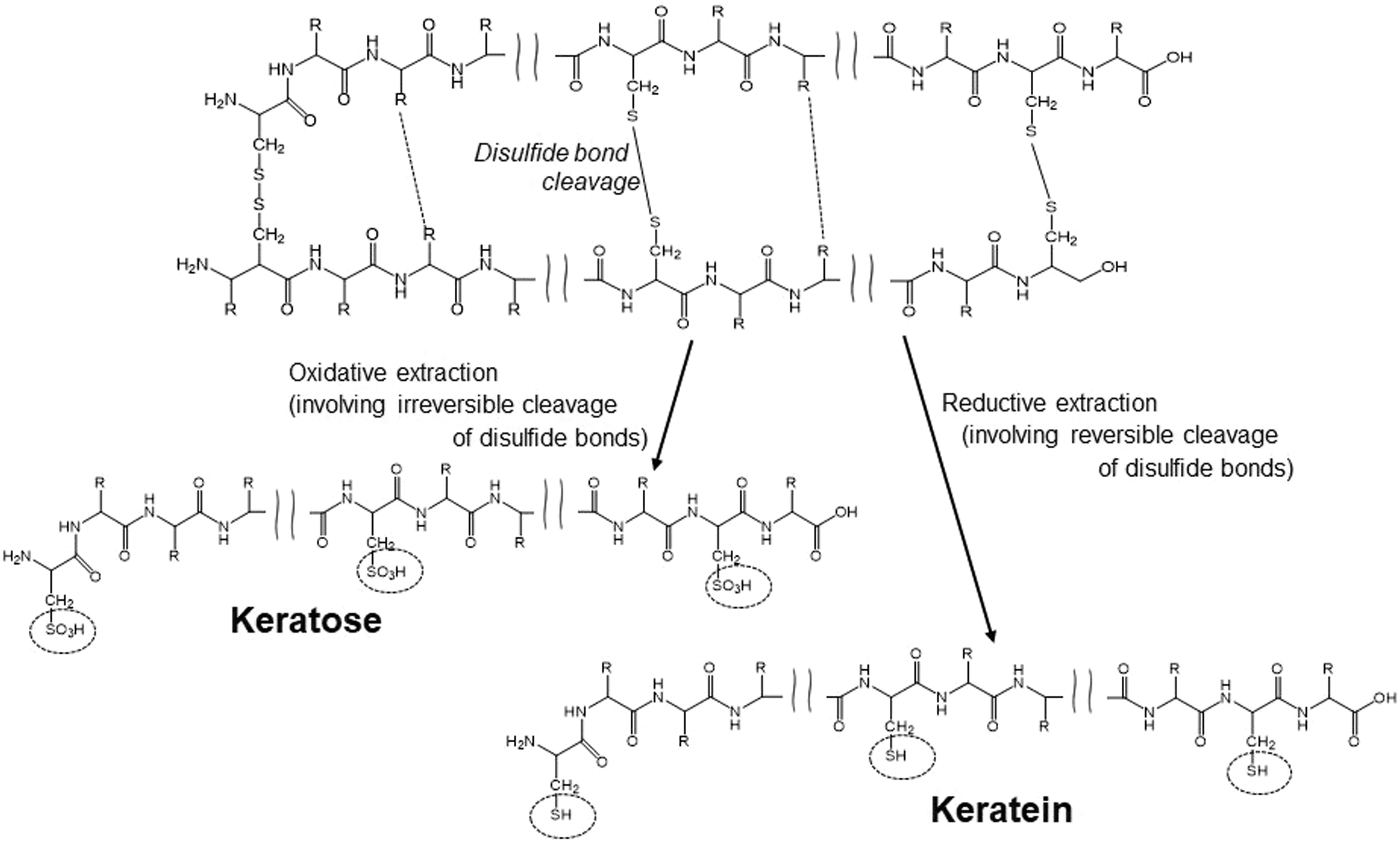
In this study, keratin obtained via reductive extraction, which retains reactive thiol (–SH) groups and is often referred to as keratein, was selectively employed to enable thiol–maleimide chemistry. For clarity, this reductively extracted keratin is hereafter referred to simply as “keratin” throughout the manuscript unless otherwise noted.
Such thiol-based reactivity offers a direct pathway for introducing functional molecules to endow keratin with specific biological properties, including anticoagulant responses. This chemical advantage expands the usability of keratin beyond traditional amino- or carboxyl-reactive biomaterials.
Heparin, a highly sulfated glycosaminoglycan, is an established anticoagulant that inhibits thrombin and prevents fibrin clot formation. It is widely applied to improve blood compatibility in vascular grafts, catheters, and other blood-contacting devices.26–28 Immobilization of heparin onto biomaterial surfaces is a proven approach to suppress coagulation; however, conventional strategies often rely on amino or carboxyl coupling, which may require harsh reagents and lead to random functionalization. 29 In addition, commonly used keratin functionalization approaches such as EDC/NHS-mediated coupling, PEGylation, and sulfation are also based on reactions involving amino or carboxyl groups, which can result in non-selective and heterogeneous modification. In contrast, maleimide–thiol coupling proceeds selectively and efficiently under physiological conditions, forming stable thiol-linked conjugates.30,31 The present study employs thiol–maleimide chemistry targeting thiol (SH) groups, enabling highly selective and controlled surface functionalization. Furthermore, the use of reductively extracted keratin rich in thiol groups provides a unique reactive platform not accessible in conventional keratin materials. This strategy enables stable immobilization of heparin with minimal release, which is rarely achieved in conventional heparinization approaches. Therefore, this study presents a promising strategy for designing keratin-based anticoagulant biomaterials.
In this study, we developed human hair–derived keratin films functionalized with heparin through maleimide–thiol coupling using an aminoethyl maleimide (AEM) linker. Keratin was extracted reductively with guanidine hydrochloride and tris(hydroxypropyl)phosphine (THPP) to preserve thiol groups, and the resulting solution was cast into thin films and cross-linked for stability. The films were subsequently reacted with heparin/AEM conjugates under mild aqueous conditions. The immobilization and surface presentation of heparin were evaluated by uronic acid assays and toluidine blue colorimetric analysis, while the resulting heparin-functionalized keratin films were further characterized spectroscopically and evaluated for anticoagulant response using thrombin-induced clotting assays. This thiol-specific immobilization strategy broadens the chemical versatility of keratin beyond conventional amino- and carboxyl-based chemistries, providing a robust, sustainable, and animal-free platform for anticoagulant surface modification of blood-contacting biomaterials.
Materials and methods
Materials
Human hair was obtained from Biolux Co., Ltd (Japan) and used as the source material for keratin extraction. All chemicals were of analytical grade and used as received without further purification. Tris (hydroxypropyl)phosphine (THPP; Nippon Chemical Industrial Co., Ltd, Japan) was employed as the reducing agent, and guanidine hydrochloride (Thermo Fisher Scientific Inc., USA) was used as the denaturant for keratin solubilization. Ellman’s reagent (5,5′-dithiobis (2-nitrobenzoic acid); DTNB) was purchased from Dojindo Laboratories (Japan) and used for the quantification of free thiol (–SH) groups. Heparin sodium salt was obtained from Nacalai Tesque, Inc. (Japan). The heterobifunctional linker N-(2-aminoethyl)maleimide hydrochloride (AEM) and the coupling reagent 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) were purchased from Combi-Blocks, Inc. (USA). Glutaraldehyde (GA) and all other solvents were supplied by FUJIFILM Wako Pure Chemical Corporation (Japan). Pellethane® segmented polyurethane (The Lubrizol Corporation, USA) was used as a comparative control material for blood-contacting experiments.
Extraction of keratin from human hair
Human hair fibers were first cleaned to remove surface lipids and contaminants. The samples were sequentially washed five times (30 min each) with 0.1% (w/v) aqueous Triton X-100, followed by five washes in a 1:4 (v/v) chloroform/methanol mixture under ultrasonic agitation. The cleaned hair fibers were then thoroughly rinsed with distilled water, air-dried at room temperature, and cut into approximately 5 mm segments.
Keratin extraction was carried out via a reductive process using 5 M guanidine hydrochloride as a denaturant and 4 wt% tris(hydroxypropyl)phosphine (THPP) as a reducing agent. The pretreated hair fragments were immersed in 200 mL of extraction solution and incubated at 50°C for 48 h under gentle stirring. After extraction, insoluble residues were removed by filtration, and the filtrate was mixed with one-third volume of chloroform to remove residual hydrophobic impurities. The aqueous phase was collected, dialyzed against distilled water for 3 days using cellulose dialysis tubing (Cellulose Tube 24/32; Sekisui Material Solutions Co., Ltd, Japan), and lyophilized to obtain purified keratin powder.
The reductive action of THPP enabled cleavage of disulfide (S–S) bonds while minimizing degradation of the keratin polypeptide backbone, resulting in keratin containing reactive thiol (–SH) groups suitable for subsequent chemical conjugation.
Molecular weight determination
The molecular weight distribution of the extracted keratin was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) under reducing conditions using β-mercaptoethanol. Keratin samples were prepared at a concentration of 3 mg/mL in sample buffer containing 2% (w/v) SDS, 10% (v/v) glycerol, 0.002% (w/v) bromophenol blue, 5% (v/v) β-mercaptoethanol, and 62.5 mM Tris–HCl (pH 6.8).
Twenty microliters of each sample were loaded per lane onto a 10% resolving gel with a 4.5% stacking gel. Electrophoresis was performed at a constant current of 20 mA for 2 h using a Mini-PROTEAN cell (Bio-Rad Laboratories, USA).
After electrophoresis, the gel was stained with Coomassie Brilliant Blue (CBB) for 1 h and destained with a methanol/acetic acid (40/10, v/v) aqueous solution until clear protein bands were visible. Molecular weight markers (PM1600; Smobio Technology, Inc., Taiwan) were used as references, and the molecular weights of keratin components were estimated by comparison with the migration distances of the standards.
The use of reducing SDS–PAGE allowed dissociation of disulfide-linked keratin aggregates into their monomeric subunits, enabling evaluation of the molecular weight distribution corresponding to type I (acidic) and type II (basic-to-neutral) keratin components.
Preparation and crosslinking of keratin films
Keratin films were prepared by a solution-casting method. Purified keratin powder was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at a concentration of 2 wt%, and the total mass of the keratin/HFIP solution was adjusted to approximately 5 g. The resulting solution was poured into 50 mm diameter Teflon-coated dishes and allowed to dry at room temperature to form uniform thin films.
For crosslinking, two different reagents—glutaraldehyde (GA) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC)—were employed under controlled aqueous conditions.
GA crosslinking
Films (8 mm diameter, 10 pieces) were immersed in 1% (v/v) GA prepared in phosphate-buffered saline [PBS(−)]. The reaction was carried out overnight (∼12 h) at 25°C with gentle shaking to ensure uniform exposure.
EDC/NHS crosslinking
For EDC-mediated crosslinking, EDC (80 mg, 0.08 g) and N-hydroxysuccinimide (NHS; 20 mg, 0.02 g) were dissolved in 10 mL of 0.05 M 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 5.6). Keratin films (8 mm diameter, 10 pieces placed in a 50 mm diameter dish) were completely immersed in this solution and gently agitated at 25°C overnight (∼12 h).
In both treatments, the films were washed five times with distilled water (DW) to remove unreacted reagents, followed by solvent exchange with ethanol and air-drying at room temperature.
The resulting crosslinked keratin films were evaluated for residual thiol (–SH) groups using Ellman’s reagent (5,5′-dithiobis (2-nitrobenzoic acid); DTNB). Each film was immersed in DTNB solution (0.1 M phosphate buffer, pH 8.0), and the absorbance of the generated 2-nitro-5-thiobenzoic acid (TNB) was measured at 412 nm using a UV–Vis spectrophotometer. Thiol concentrations were determined using a cysteine calibration curve (R2 ≥ 0.99).
This assay enabled quantitative evaluation of reactive thiol groups retained after GA and EDC crosslinking under defined conditions.
Synthesis of heparin/AEM conjugate
Heparin was functionalized with N-(2-aminoethyl)maleimide hydrochloride (AEM) through carbodiimide-mediated coupling to obtain a maleimide-terminated intermediate suitable for subsequent thiol–maleimide conjugation (Figure 2, lower panel). Preparation of keratin films and schematic illustration of heparin immobilization. Top: formation and crosslinking of keratin films. Bottom: maleimide–thiol conjugation between keratin thiol (–SH) groups and the heparin/AEM complex through Michael-type addition.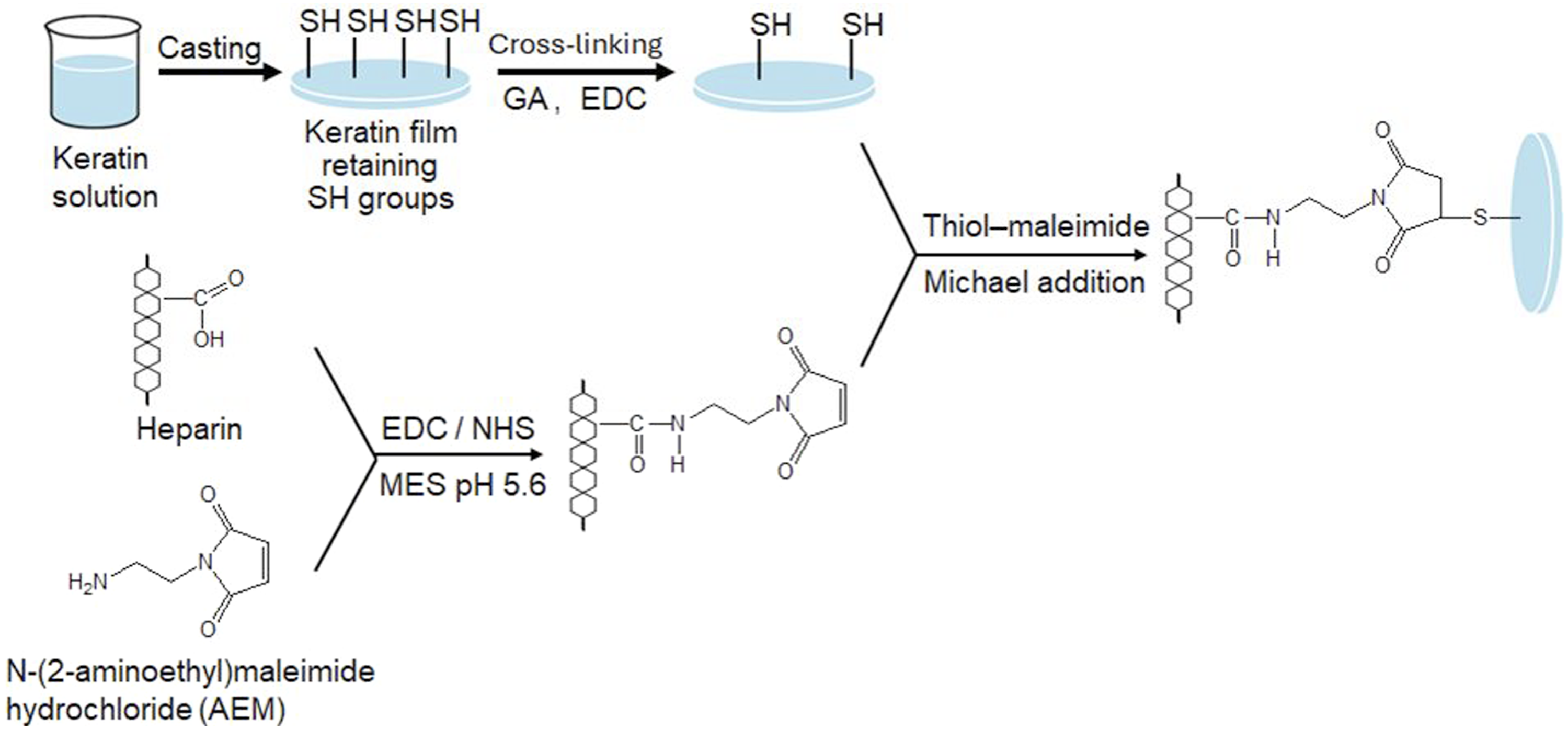
Briefly, heparin (10 mg/mL) and AEM were reacted in 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 5.6) in the presence of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) for 12 h at room temperature. The initial feed ratio of AEM to heparin was approximately 57:1 (mol/mol), calculated based on an assumed average molecular weight of heparin (12,000).
The reaction mixture was purified by dialysis (molecular weight cutoff: 12–16 kDa) at 4°C against distilled water with an exchange volume ratio of ≥1:100, using 6–8 buffer changes over 72 h. The purified retentate (heparin/AEM conjugate) was lyophilized and stored in a desiccated state at 4°C.
The chemical structure of the conjugate was confirmed by attenuated total reflection–Fourier transform infrared (ATR–FTIR) spectroscopy (FT/IR-4200, JASCO Corp., Japan). The spectra were analyzed for the appearance of characteristic amide I (∼1650 cm−1) and amide II (∼1540 cm−1) bands, indicating successful amide bond formation between heparin and AEM.
Immobilization of heparin onto keratin films
The heparin/AEM conjugate was immobilized onto keratin films through maleimide–thiol coupling (Michael addition reaction), as illustrated in Figure 2, which depicts the overall reaction sequence from AEM functionalization of heparin to its covalent attachment to keratin. Dried keratin films were immersed in a heparin/AEM solution prepared in MES buffer (pH 7.0) and incubated for 6 h at room temperature. During this process, the maleimide groups of AEM selectively reacted with thiol (–SH) groups of keratin, resulting in heparin–keratin conjugation through thiol–maleimide coupling.
After the reaction, the films were thoroughly rinsed with buffer to remove unbound components and subsequently air-dried under ambient conditions.
The extent of heparin conjugation was evaluated using two complementary approaches: (1) quantification of residual thiol groups remaining in the keratin films using Ellman’s assay, and (2) determination of the decrease in heparin concentration in the reaction solution using the carbazole–sulfuric acid assay, which is specific for uronic acid residues.
To assess potential heparin release, heparin/AEM-immobilized keratin films (8 mm diameter) were immersed in PBS (pH 7.4) at 37°C for 24 h and 7 days. At each time point, soaking media were collected and analyzed for uronic acid content using the carbazole–sulfuric acid method as described above. Heparin concentrations were calculated from a sodium heparin calibration curve (0–50 µg/mL, R2 ≥ 0.99), with a detection limit of approximately 1 µg/mL (blank +3 SD). Spike recovery tests using heparin-spiked PBS samples showed 95–105% recovery (n = 3). As a positive control, the immobilization reaction supernatant was quantified in parallel to confirm assay performance.
No detectable heparin (<LOD) was detected in the soaking media at any time point.
Toluidine blue staining for surface heparin verification
The presence of surface-immobilized heparin on keratin films was evaluated by toluidine blue (TB) staining, as a qualitative/semi-quantitative indicator of surface-bound heparin. Toluidine blue is known to electrostatically interact with sulfated glycosaminoglycans. Unmodified keratin films, heparin/AEM-immobilized keratin films, and Pellethane® polyurethane films were prepared as described above.
Samples were immersed in 0.05% (w/v) toluidine blue O solution prepared in 0.01 M sodium phosphate buffer (pH 6.8) for 30 min at room temperature. After staining, the films were gently rinsed with distilled water until the wash solution became colorless and then air-dried under ambient conditions.
To evaluate surface-bound TB without dye extraction, solid-state colorimetric measurements were performed using a spectrocolorimeter (NF-333, Nippon Denshoku Ind. Co., Ltd, Japan). Measurements were conducted under illuminant D65 with a 10° standard observer and an 8 mm measurement aperture. Samples were placed on an opaque neutral-gray backing plate (Munsell N6) to minimize background effects.
Color coordinates (L*, a*, b*) in the CIELAB color space were recorded after TB staining. The b* value was used as a quantitative indicator of surface-bound heparin, because TB binding to sulfate groups results in increased blue coloration on the film surface (lower b* values). Measurements were performed at 10 different positions per film, with three independent films analyzed per group. Data are presented as mean ± standard deviation. Statistical analysis was conducted using one-way ANOVA followed by Tukey’s post hoc test, with statistical significance defined as *p < 0.05.
Surface characterization of heparin-functionalized keratin films
To further confirm the presence of heparin on the keratin film surface, X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FT-IR) analyses were performed for untreated keratin films and heparin/AEM-immobilized keratin films.
XPS measurements were conducted using an AXIS-ULTRA DLD X-ray photoelectron spectrometer (KRATOS Analytical Ltd, Japan) equipped with a monochromatic Al Kα X-ray source (hν = 1486.6 eV). High-resolution spectra were acquired in the S 2p region with a pass energy of 20 eV to analyze sulfur-containing species on the film surfaces.
The binding energy scale was calibrated using the C 1s peak at 284.8 eV as a reference. The presence of sulfate groups (–SO3−) derived from immobilized heparin was evaluated based on the characteristic S 2p binding energy region around 168–170 eV. Relative changes in sulfur-related signals were compared between untreated keratin films and heparin-functionalized keratin films.
FT-IR measurements were performed according to the procedure described in the Materials and Methods section using an attenuated total reflectance (ATR) mode. FT-IR analysis here focuses on sulfate-related bands distinct from the amide bond formation discussed in the Materials and Methods section Spectra were recorded to analyze the chemical structure of the film surfaces.
The presence of heparin was evaluated by identifying characteristic absorption bands attributed to sulfate groups (–SO3−), including the asymmetric S = O stretching vibration around 1220–1260 cm−1 and the symmetric S = O stretching vibration around 1030–1060 cm−1. Particular attention was given to the band at 1030–1060 cm−1, which provides a more reliable indication due to minimal overlap with intrinsic keratin signals.
The spectra were compared between untreated and heparin-functionalized films to assess changes associated with the introduction of sulfate functionalities.
Anticoagulant activity assay
A thrombin-induced coagulation assay was employed to evaluate the anticoagulant response of free and immobilized heparin. Commercially available citrated porcine plasma was used for all experiments. Plasma was equilibrated on ice prior to use.
For each measurement, 200 μL of plasma was mixed with 200 μL of thrombin solution (5 U/mL; FUJIFILM Wako Pure Chemical Corporation, Japan). The absorbance at 405 nm was continuously monitored using a UV–Vis spectrophotometer (UV-1900i, Shimadzu Corp., Japan) to track fibrin formation in real time.
The following conditions were evaluated: (i) plasma containing free heparin or heparin/AEM complex (homogeneous system), (ii) plasma in contact with heparin-immobilized keratin films (heterogeneous system), and (iii) plasma in contact with unmodified keratin films or Pellethane® as control materials.
The coagulation rate was determined from the initial slope of the absorbance–time curve. The thrombin-only condition was normalized to 1.0 as a reference for comparison among groups.
Scanning Electron Microscopy (SEM) observation
The surface morphology of keratin films was examined using scanning electron microscopy (SEM). Samples were mounted on aluminum stubs using conductive carbon tape and sputter-coated with a thin platinum layer (JFC-1600, JEOL Ltd, Tokyo, Japan) to prevent surface charging during observation.
SEM observations were performed using a JSM-6390LM microscope (JEOL Ltd, Japan) under high-vacuum conditions at an accelerating voltage of 10 kV. Images were acquired at a magnification of ×300.
For each sample (untreated, crosslinked, and heparin/AEM-immobilized), at least three different regions were observed to ensure reproducibility. Representative images were selected from these observations to compare surface morphology and film continuity.
Particular attention was paid to evaluating surface smoothness, the presence of defects or phase separation, and any structural changes associated with crosslinking and subsequent heparin immobilization.
Water contact angle measurement
The surface wettability of the keratin films was evaluated by static water contact angle measurements using a contact angle goniometer (FTA125 Contact Angle Analyzer, First 10 Angstroms, USA). A droplet of ultrapure water (5 μL) was carefully deposited onto the film surface using a microsyringe.
The contact angle was measured immediately after droplet deposition using the sessile drop method. All measurements were conducted under controlled environmental conditions (25°C and 50% relative humidity).
For each sample, contact angles were measured using six independently prepared films (n = 6), with each measurement performed on a separate film to ensure statistical independence. The reported values represent the mean ± standard deviation.
Statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test, with differences considered statistically significant at *p < 0.05.
Mechanical testing
The tensile properties of keratin films were evaluated by uniaxial tensile testing to assess the effects of crosslinking and subsequent heparin immobilization on mechanical integrity. Film specimens prepared by the casting method were cut into rectangular strips (5 mm in width and 25 mm in length).
Mechanical testing was carried out using a universal testing machine (EZ-LX, Shimadzu Corp., Kyoto, Japan) at a constant crosshead speed of 2 mm min−1. To ensure reproducibility, specimens were randomly sampled from multiple independently prepared films for each condition (n = 3–4).
Prior to testing, all samples were equilibrated under controlled environmental conditions (25°C, 40% relative humidity) for at least 48 h. Tensile measurements were conducted under the same conditions to minimize the influence of moisture on mechanical behavior.
Stress–strain curves were obtained from the recorded load–displacement data. Tensile stress was calculated using the initial cross-sectional area of each specimen, and strain was determined by normalizing displacement with respect to the initial gauge length.
The apparent Young’s modulus was calculated from the slope of the initial linear region (ε = 0–0.02) of the stress–strain curve. The tensile strength was defined as the maximum stress obtained from the stress–strain curve.
Results and discussion
Overview of experimental design
To provide a comprehensive understanding of the fabrication and evaluation process, the experimental workflow of this study consisted of four consecutive stages: (1) extraction and characterization of keratin from human hair, (2) preparation and crosslinking of keratin films, (3) covalent immobilization of heparin via maleimide–thiol coupling, and (4) evaluation of anticoagulant response.
Particular emphasis was placed on verifying the preservation and utilization of thiol (–SH) groups for selective chemical conjugation, as well as correlating surface modification with anticoagulant functionality. Each stage was systematically analyzed to confirm the successful integration of these processes and their contribution to the overall performance of the heparin-functionalized keratin films.
Extraction and characterization of keratin
Keratin was extracted using a reductive system comprising 5 M guanidine hydrochloride and 4 wt% tris(hydroxypropyl)phosphine (THPP). Compared with the conventional urea/2-mercaptoethanol method, this approach yielded a higher extraction efficiency (42.6% vs 10–20%).31,32 This improvement can be attributed to the combined effect of the strong denaturing capability of guanidine hydrochloride and the mild yet selective reducing ability of THPP.
Whereas 2-mercaptoethanol is known to induce extensive reduction and fragmentation of keratin chains under certain conditions,33,34 THPP allows more controlled cleavage of disulfide bonds while maintaining oxidative stability. As a result, molecular degradation is reduced and a higher proportion of reactive thiol groups is retained, which is advantageous for subsequent conjugation reactions.
SDS–PAGE analysis (Figure 3) revealed distinct bands at approximately 40–45 kDa and 50–60 kDa, corresponding to acidic (type I) and basic-to-neutral (type II) keratins, respectively.35,36 In addition, faint bands observed in the 10–30 kDa range were attributed to keratin-associated proteins (KAPs) or γ-keratins, which are characteristic components of α-keratin Ref. 37. These banding patterns indicate that the extraction process preserved both major keratin subunits and minor associated proteins, resulting in a molecular weight distribution comparable to that of native human hair keratin. Molecular weight profile of extracted keratin (SDS–PAGE). SDS–PAGE analysis showing type I and type II keratin bands together with minor keratin-associated proteins characteristic of native human hair keratin.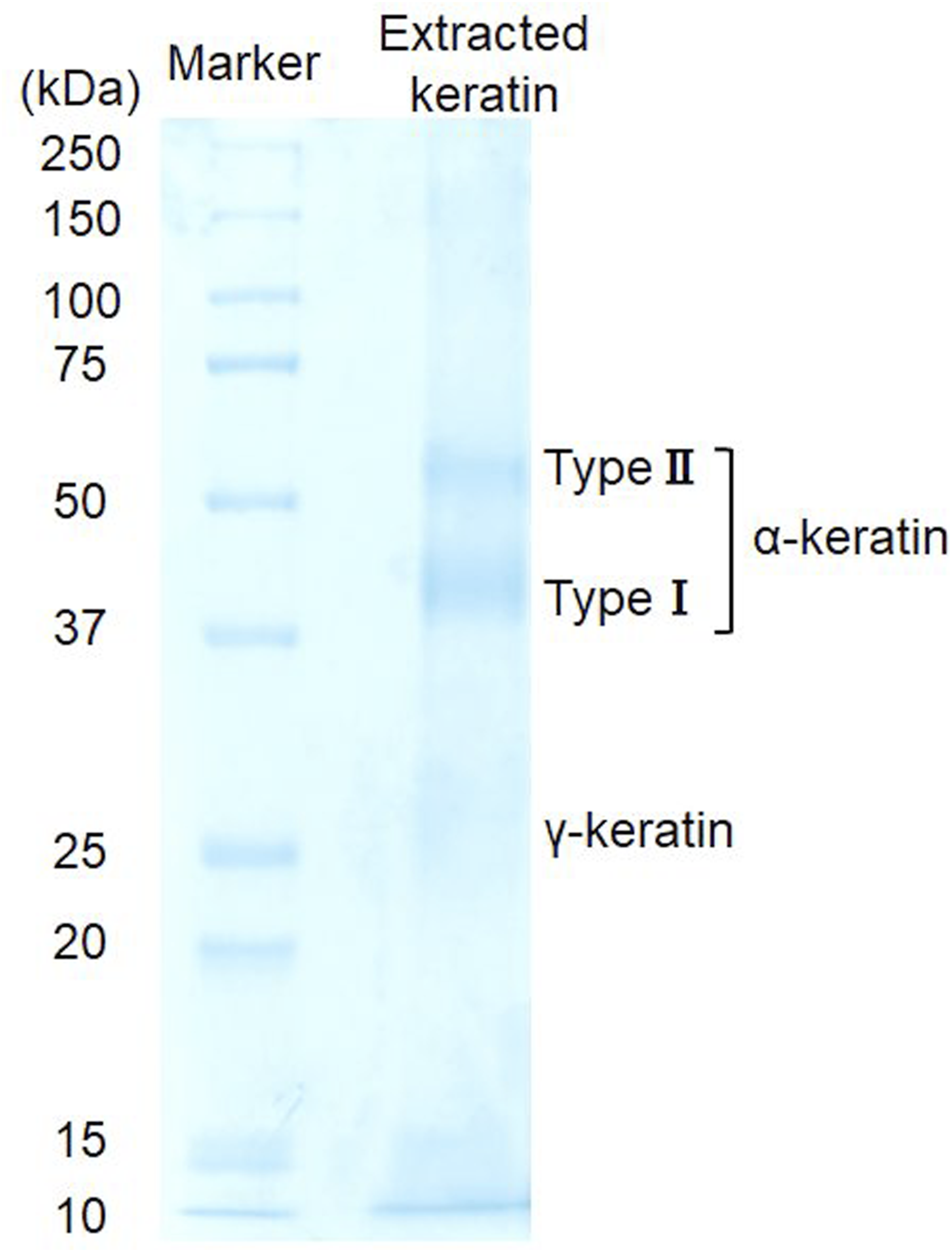
Overall, the guanidine/THPP system enabled efficient solubilization of keratin while limiting excessive protein degradation relative to conventional reductive methods. This reductive extraction approach therefore provides a reliable means of obtaining thiol-functionalized keratin suitable for subsequent chemical modification and biomaterial fabrication.
Quantification of residual thiol groups
The preservation of thiol (–SH) groups after extraction and film formation was quantified using Ellman’s reagent. As shown in Figure 4, keratin films exhibited a clear mass-dependent increase in SH content, whereas gelatin films—used as a thiol-deficient control—showed no detectable absorbance. This result indicates that background interference in the assay was minimal and that the extracted keratin retained a substantial amount of free thiol groups after purification and film casting. Quantification of residual thiol groups on keratin films. Ellman’s reagent assay comparing residual thiol (–SH) contents in keratin films before and after crosslinking with glutaraldehyde (GA) or EDC. Data are presented as mean ± SD. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparison test. ***p < 0.001 versus the corresponding GA-crosslinked film at the same keratin content.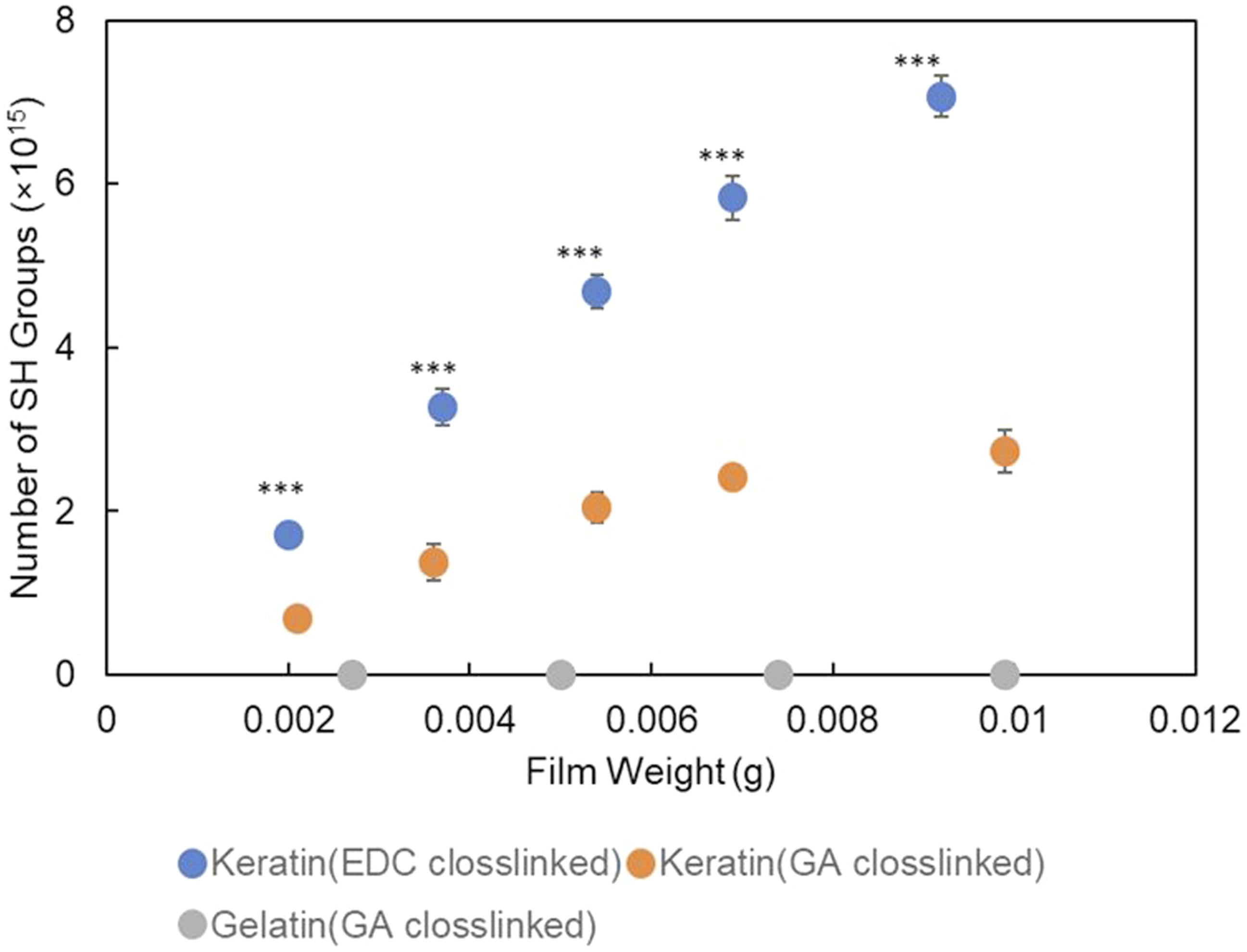
Crosslinking treatment significantly affected the residual SH content. Glutaraldehyde (GA) crosslinking led to a pronounced reduction in measurable thiol groups compared with EDC crosslinking, which can be attributed to the nonspecific reactivity of GA toward various nucleophilic sites, including thiols, through aldehyde–Schiff base formation. 38 In contrast, EDC primarily activates carboxyl groups to form amide bonds with amino residues, thereby largely preserving thiol functionalities. This selective reactivity is advantageous compared to glutaraldehyde-based crosslinking, which may lead to partial consumption or modification of thiol groups through thiol–aldehyde interactions such as thiohemiacetal formation.
Based on these observations, EDC-crosslinked keratin films were selected for subsequent heparin conjugation experiments, as they retained a higher density of reactive thiol groups, which is a key prerequisite for efficient maleimide–thiol coupling during the immobilization process.
Formation and characterization of heparin/AEM complex
To enable thiol-specific immobilization, heparin was reacted with N-(2-aminoethyl)maleimide (AEM) in the presence of EDC to obtain a maleimide-functionalized heparin derivative. The reaction scheme is shown in Figure 2 (lower panel). The formation of the heparin/AEM conjugate was examined by ATR–FTIR spectroscopy (Figure 5). The appearance of absorption bands at approximately 1650 cm−1 (amide I, C = O stretching) and 1540 cm−1 (amide II, N–H bending and C–N stretching) is consistent with the formation of amide linkages between the carboxyl groups of heparin and the amino group of AEM.39,40 In parallel, a decrease in the intensity of carboxylate-related bands originating from heparin uronic acid residues was observed, accompanied by the emergence of amide I and II bands, supporting carbodiimide-mediated amide bond formation rather than simple physical association. FT-IR spectra of heparin/AEM complex. ATR-FTIR spectra showing newly appeared amide I (∼1650 cm−1) and amide II (∼1540 cm−1) bands after conjugation of AEM to heparin, while characteristic sulfate-related absorption bands derived from heparin remained preserved.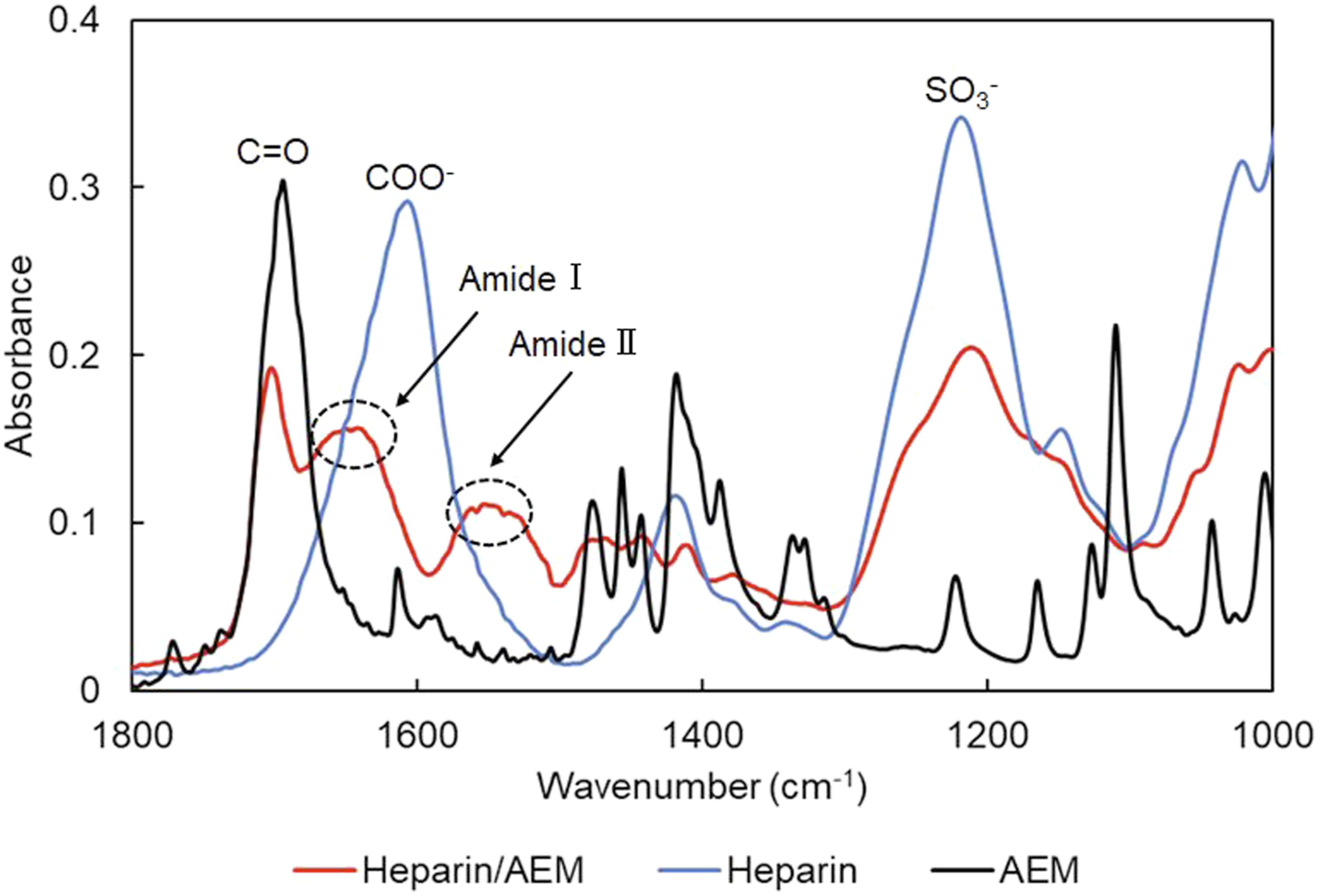
In addition, the characteristic sulfate absorption band around 1250 cm−1 was retained, indicating that the sulfated polysaccharide backbone of heparin remained intact after conjugation.
These results suggest the successful introduction of AEM onto the heparin backbone, although direct spectroscopic identification of the maleimide moiety is limited due to overlap with neighboring absorption bands.
Taken together, these spectral features indicate that AEM was successfully introduced onto the heparin backbone, providing maleimide functionality suitable for subsequent thiol–maleimide coupling while preserving the essential structural characteristics of heparin.
Anticoagulant activity of the heparin/AEM complex
The anticoagulant activity of the heparin/AEM complex was evaluated using a thrombin-induced plasma clotting assay. This assay was selected because it allows direct evaluation of surface-mediated anticoagulant activity, whereas conventional APTT and PT assays primarily reflect bulk-phase coagulation responses. In particular, this assay enables direct assessment of fibrin formation at the material–plasma interface, which is critical for evaluating anticoagulant performance of surface-immobilized heparin. As shown in Figure 6(a), rapid fibrin formation was observed in the thrombin-only control, whereas plasma supplemented with either free heparin or the heparin/AEM complex showed no appreciable increase in absorbance within the measurement period, indicating effective suppression of coagulation. The clotting time, defined as the time required to reach 50% of the maximum absorbance, was approximately 90 s for the thrombin-only control. In contrast, no detectable clot formation was observed for either free heparin or the heparin/AEM complex within the measurement period. Anticoagulant activity of the heparin/AEM complex in thrombin-induced plasma clotting. (a) Time-dependent fibrin formation monitored by absorbance changes at 405 nm. (b) Relative coagulation rates calculated from the initial slope of the absorbance–time curves, showing comparable anticoagulant activity between free heparin and the heparin/AEM complex.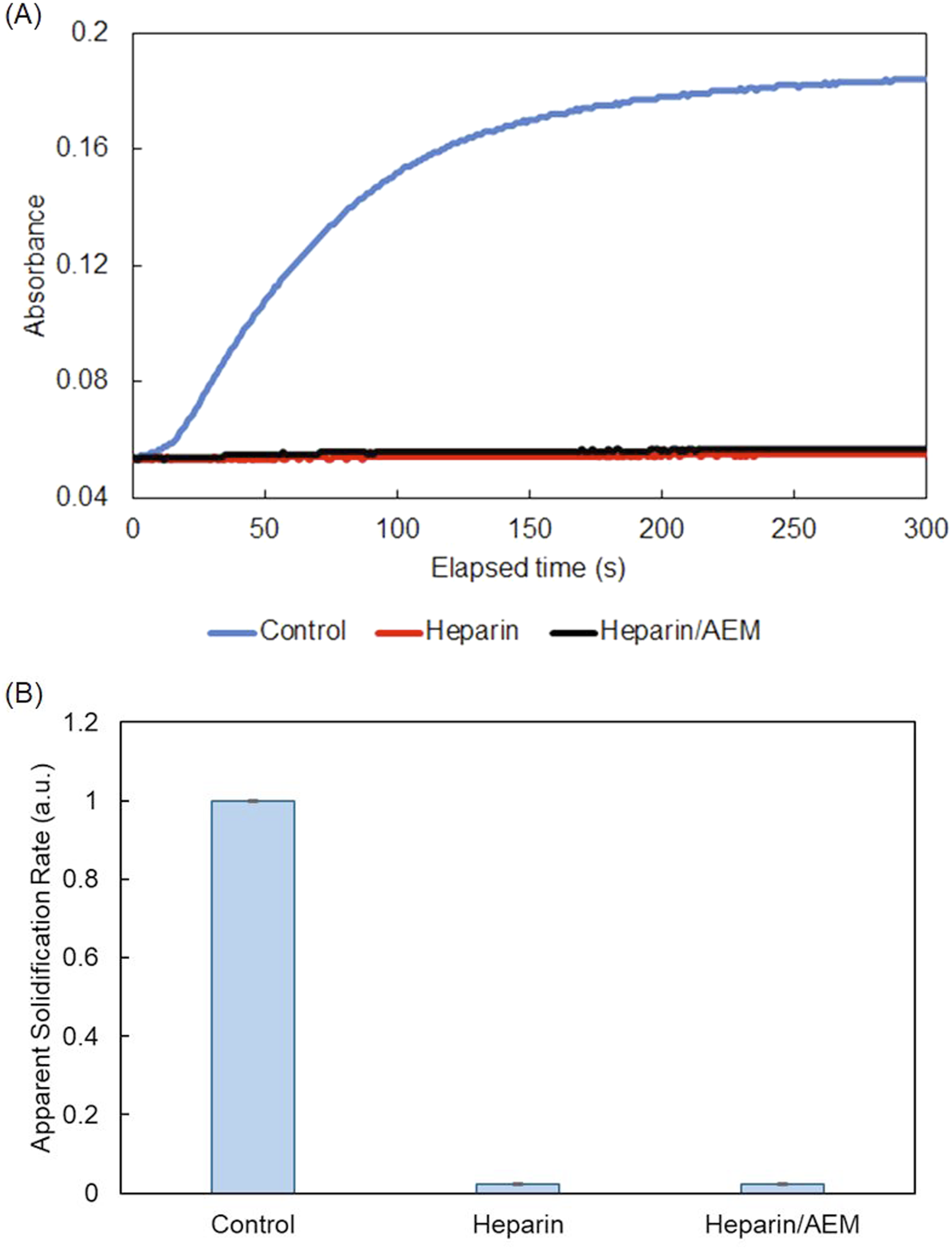
Quantitative analysis of the initial clotting rate (Figure 6(b)) further showed that both heparin-containing samples exhibited comparable inhibitory effects, markedly reducing the apparent clotting rate relative to the control condition.
These results indicate that conjugation of heparin with AEM does not substantially impair its anticoagulant function, supporting the suitability of this maleimide-functionalized heparin for subsequent covalent immobilization onto keratin-based biomaterial surfaces.41,42
Immobilization of heparin onto keratin films
Heparin/AEM conjugates were covalently immobilized onto keratin films via maleimide–thiol addition, a selective and biocompatible coupling reaction.43,44 The immobilization process was evaluated using two complementary analyses: (1) quantification of thiol consumption within the keratin films, and (2) determination of heparin depletion in the reaction medium.
As shown in Figure 7, the amount of residual –SH groups in the keratin films decreased proportionally with increasing heparin/AEM concentration, indicating progressive consumption of thiols during covalent coupling. Consumption of thiol groups on keratin films following heparin immobilization. Decrease in measurable thiol (–SH) groups after reaction with the heparin/AEM complex, supporting thiol–maleimide coupling between keratin and the heparin/AEM complex. Statistical analysis was performed using one-way ANOVA followed by Dunnett’s test. *p < 0.01 and **p < 0.001 versus 0 µg/mL.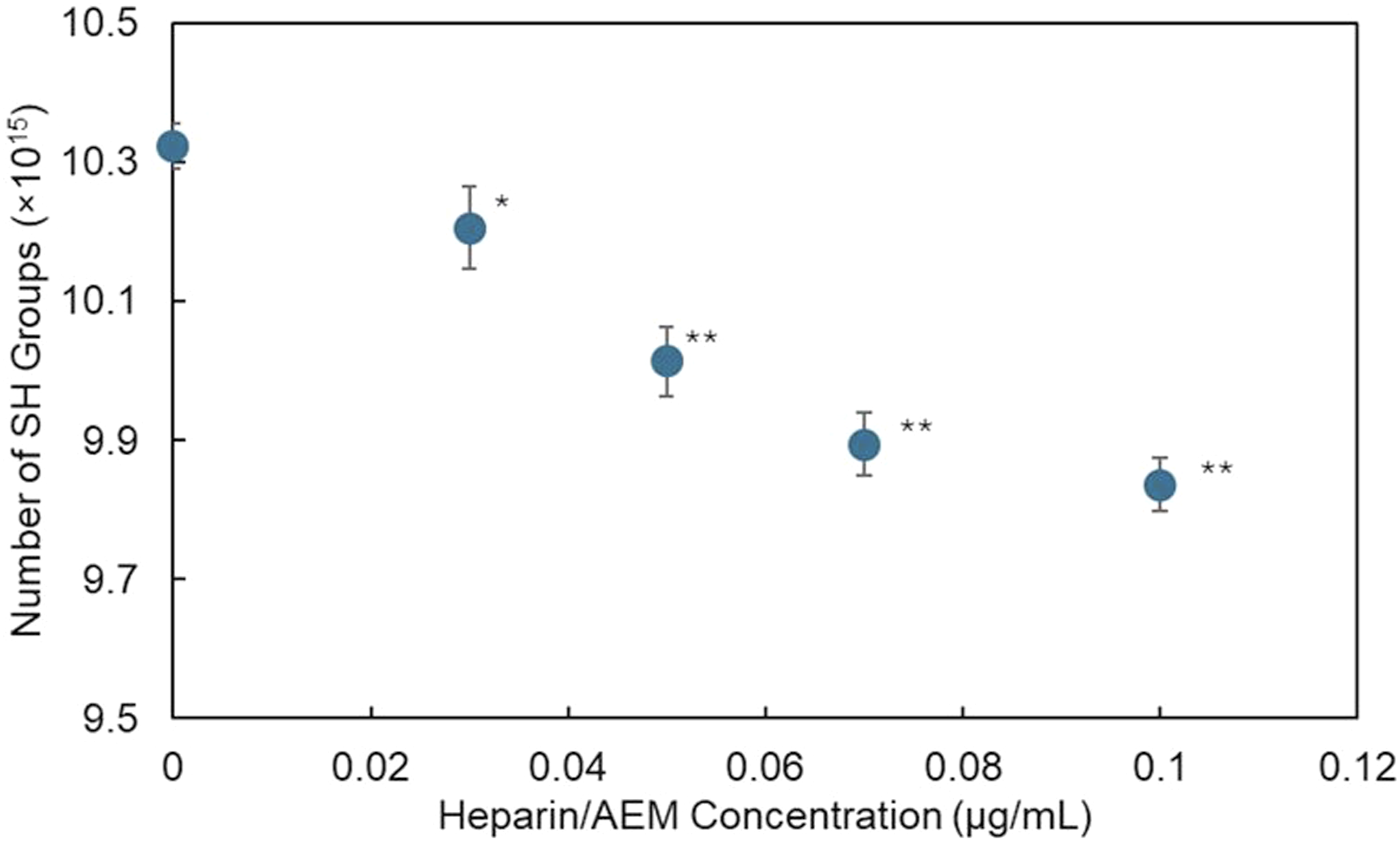
In parallel, the concentration of heparin remaining in the reaction solution significantly decreased after immobilization, as shown in Figure 8, further supporting successful immobilization onto the keratin films. Decrease in heparin concentration in solution during immobilization. Reduction in uronic-acid concentration in the reaction medium before and after immobilization, demonstrating depletion of heparin from solution due to immobilization onto keratin films.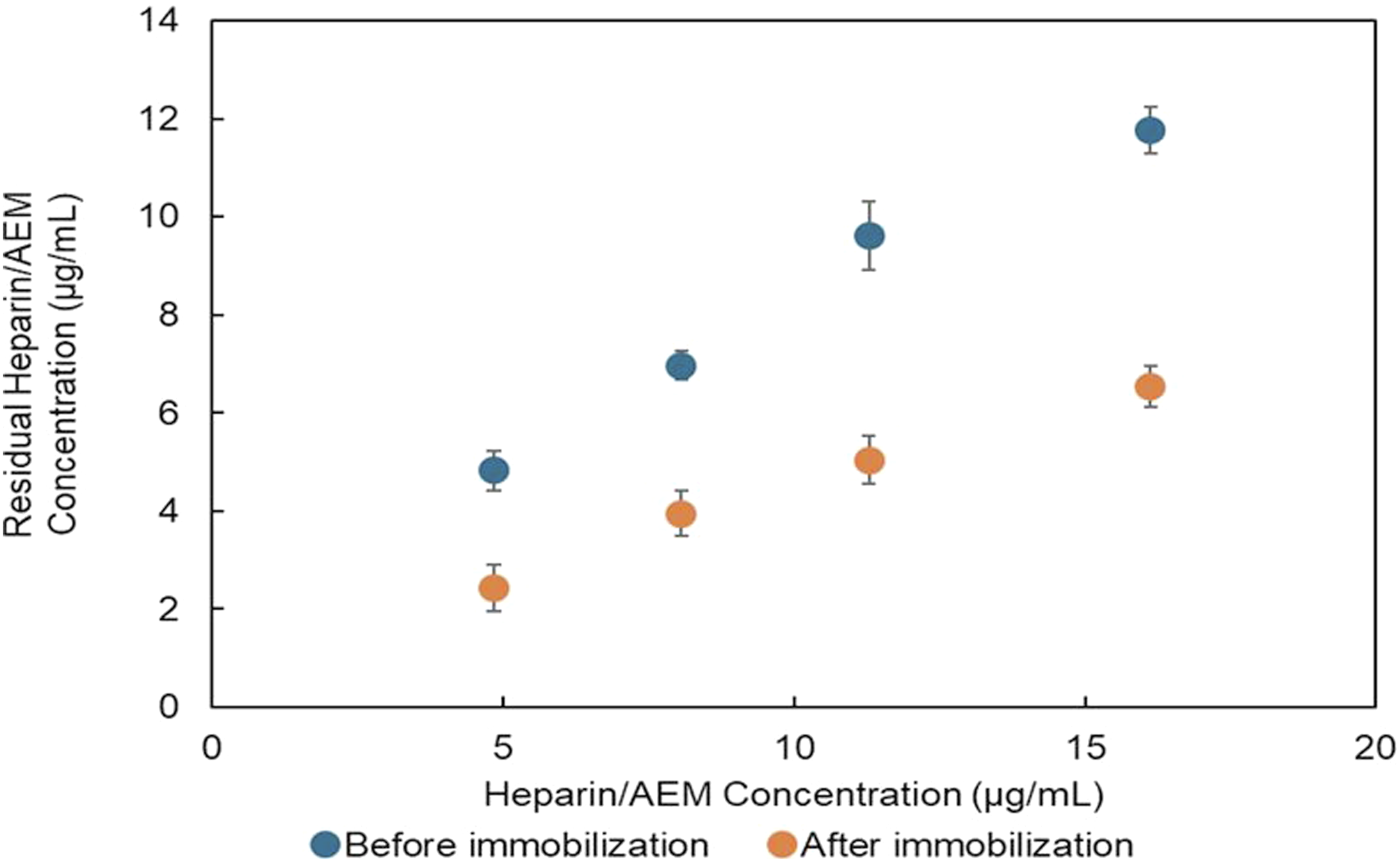
The accessible surface thiol content of keratin films was quantified by Ellman’s assay, and the decrease in SH groups after reaction corresponded to approximately 0.76 nmol per film, which is equivalent to about 4.4% of the initial accessible surface thiols. This decrease in thiol content provides quantitative evidence of thiol–maleimide coupling on the keratin surface.
Based on the decrease in surface SH groups, the approximate surface density of immobilized heparin was estimated to be ∼ 0.02 nmol/cm2.
To further evaluate the stability of the immobilized heparin, the films were immersed in PBS (pH 7.4, 37°C) for 24 h and 7 days, and the soaking media were analyzed for uronic-acid residues using the carbazole–sulfuric acid assay. As shown in Figure 9, no detectable heparin (<1 µg/mL, detection limit; spike recovery 95–105%, R2 ≥ 0.99) was observed in any soaking medium, whereas the positive-control solution containing free heparin/AEM exhibited a clear absorbance at 530 nm. These results indicate that the heparin/AEM complex remained immobilized on the keratin surface without measurable release under the tested conditions. This corresponds to a theoretical upper-bound release estimate on the order of 103 ng/cm2, indicating negligible heparin release from the film surface. Release of heparin from immobilized keratin films. Quantification of released heparin in soaking buffer using the carbazole–sulfuric acid assay. No detectable heparin release was observed within the detection limit under the tested conditions.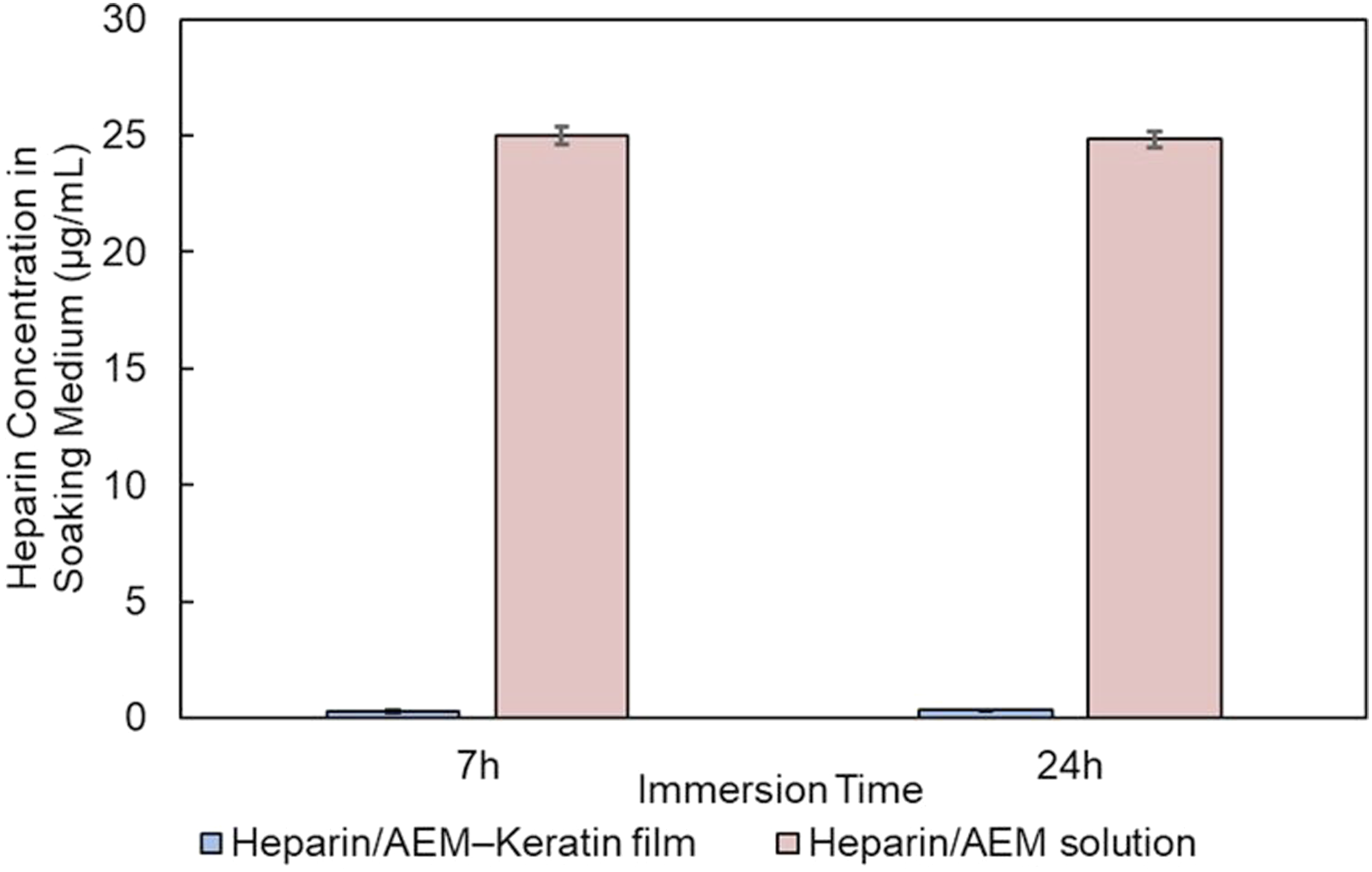
Surface presentation of immobilized heparin was further examined by toluidine blue (TB) staining followed by solid-state colorimetric analysis, as described in the Materials and Methods section. As shown in Figure 10, heparin-immobilized keratin films exhibited significantly lower b* values than unmodified keratin and Pellethane®, reflecting increased blue coloration due to sulfate-bound TB (one-way ANOVA with Tukey’s test, p < 0.05). Quantification of toluidine blue uptake on film surfaces based on CIELAB b* values. Comparison of b* values for untreated keratin, heparin/AEM-immobilized keratin, and Pellethane® films after toluidine blue staining. Lower b* values indicate stronger blue coloration and higher surface presentation of sulfated groups derived from immobilized heparin.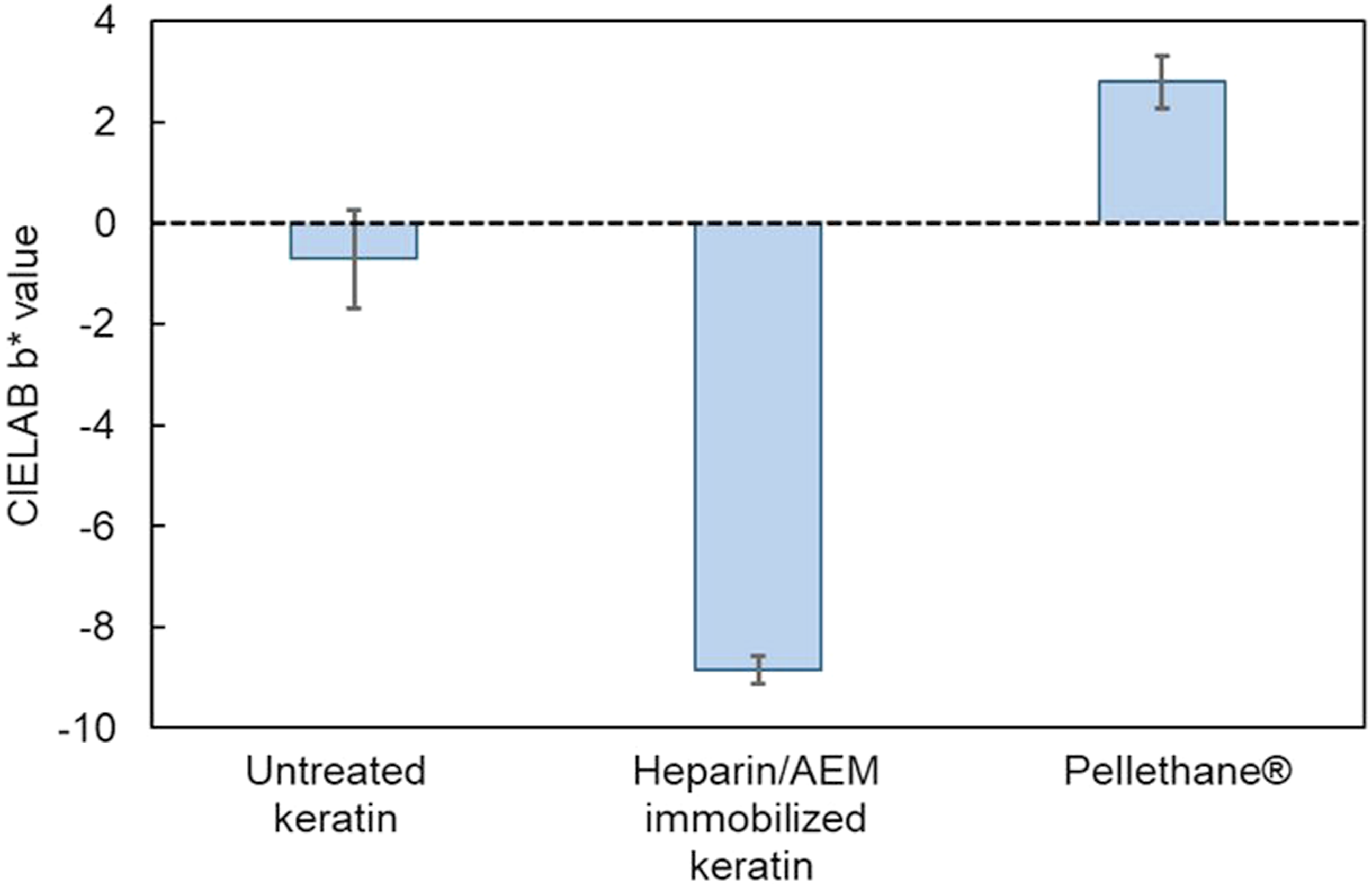
Unlike conventional TB assays based on dye extraction, the solid-state b* analysis provides a non-destructive and surface-sensitive measure of sulfated groups, enabling direct comparison across substrates with different intrinsic optical properties.
Collectively, these findings strongly support stable immobilization and surface presentation of heparin on keratin films via maleimide–thiol coupling rather than simple physical adsorption, based on the combined evidence of thiol consumption, negligible release, and surface-selective TB staining.
This multi-tier evaluation, integrating film-phase, solution-phase, and surface-colorimetric analyses, further supports stable immobilization of heparin on the keratin film surface.38,40,43,45 Compared to conventional keratin functionalization approaches such as EDC/NHS coupling, PEGylation, and sulfation, the present thiol–maleimide strategy provides improved selectivity and stable surface immobilization through thiol-specific conjugation.
Surface chemical characterization
XPS analysis was performed to evaluate the surface chemical composition of keratin films before and after heparin immobilization.
As shown in Figure 11(a), the survey spectra of the untreated keratin film mainly exhibited signals corresponding to carbon (C 1s), oxygen (O 1s), and nitrogen (N 1s), whereas the heparin-immobilized keratin film additionally showed a detectable sulfur-related signal. For clarity, the spectra were vertically offset. No remarkable changes were observed in the C 1s and N 1s regions after heparin immobilization. This is likely because both keratin and the heparin/AEM conjugate contain overlapping carbon- and nitrogen-containing structures, resulting in similar spectral profiles in these regions. Quantitative XPS analysis further revealed the appearance of sulfur in the heparin-functionalized sample, while sulfur was not detected in the untreated keratin film under the present measurement conditions (Table 1). In addition, the S/C atomic ratio increased from not detectable to 0.0087 after heparin immobilization, indicating the introduction of sulfur-containing functionalities onto the film surface. Surface chemical characterization of heparin-immobilized keratin films by XPS and FT-IR. (a) XPS survey spectra of untreated and heparin/AEM-immobilized keratin films. For clarity, the spectra were vertically offset. (b) High-resolution S 2p XPS spectra showing sulfate-derived sulfur species around 168–170 eV in the heparin/AEM-immobilized keratin film. (c) ATR-FTIR spectra showing sulfate-related absorption bands assigned to asymmetric and symmetric SO3- stretching vibrations. Surface elemental composition of keratin films determined by XPS survey analysis. Elemental composition is presented as atomic percentage (at.%). N.D.: not detected.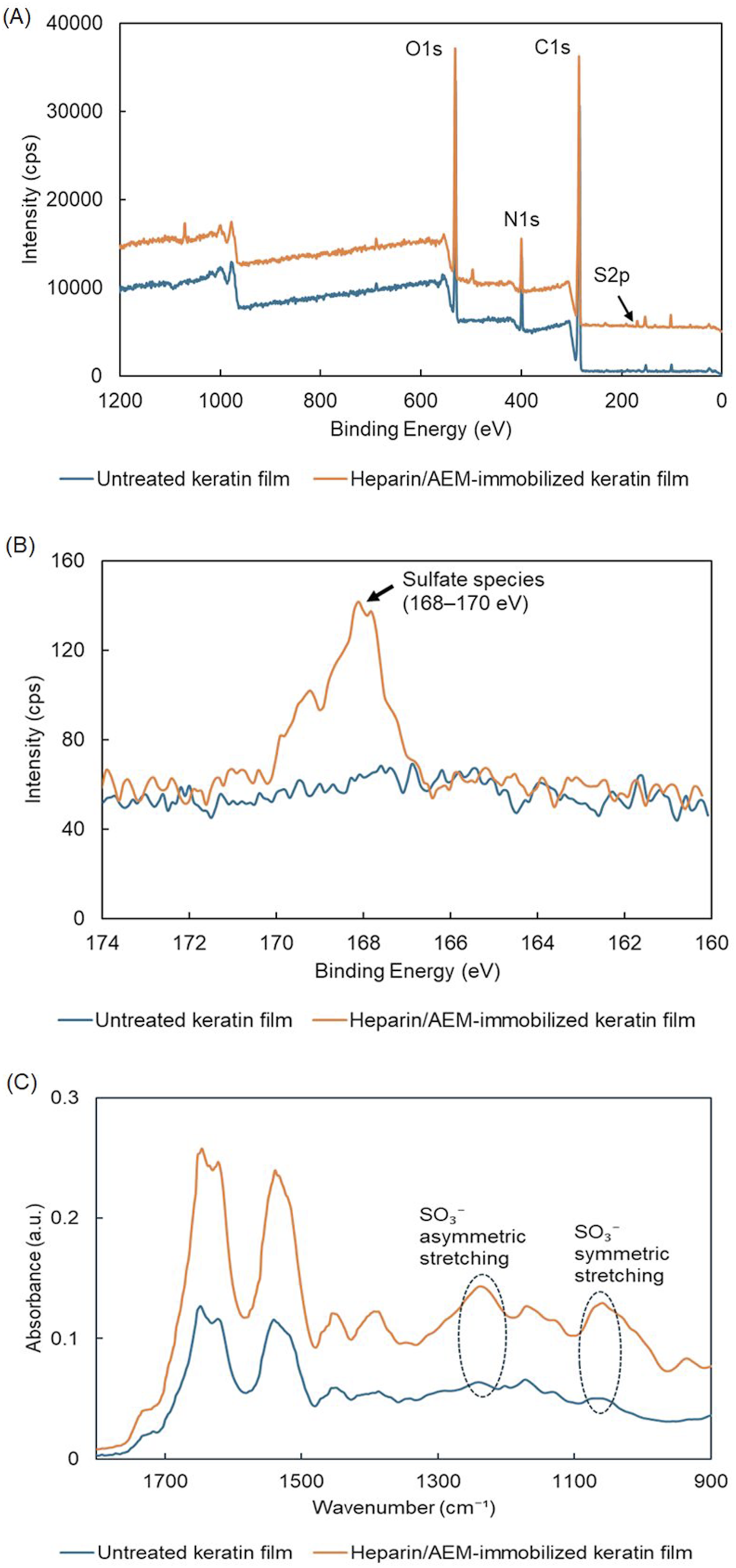

To further characterize the sulfur chemical state, high-resolution XPS spectra were recorded in the S 2p region (Figure 11(b)). The heparin-immobilized keratin film exhibited a distinct sulfate-derived S 2p signal centered around approximately 168.4 eV, which is consistent with highly oxidized sulfur species such as sulfate groups (–SO3-) derived from heparin. In contrast, untreated keratin films did not exhibit a clear signal in this binding-energy region. Since intrinsic keratin sulfur originating from thiol or disulfide groups is generally observed at lower binding energies (∼163–165 eV), the appearance of the oxidized sulfur signal strongly supports the surface presence of heparin-derived sulfate functionalities.
FT-IR analysis further supported successful heparin incorporation onto the keratin films. As shown in Figure 11(c), the heparin-immobilized keratin films exhibited characteristic sulfate-related absorption bands, including asymmetric S = O stretching vibrations around 1220–1260 cm−1 and symmetric S = O stretching vibrations around 1030–1060 cm−1. In particular, the increased absorption intensity near 1030–1060 cm−1 compared with untreated keratin films indicates the introduction of sulfate-containing structures derived from heparin.
These spectroscopic findings are consistent with the presence of sulfated glycosaminoglycan structures and cannot be attributed solely to the intrinsic keratin backbone. Furthermore, the XPS and FT-IR results complement the findings from thiol-consumption analysis, toluidine blue staining, and the absence of detectable heparin release, all of which independently support stable immobilization and surface presentation of heparin on the keratin film surface.
Overall, the combined surface-sensitive XPS analysis and complementary FT-IR characterization provide strong evidence for successful immobilization and surface presentation of heparin-derived sulfate groups on the keratin films.
Surface morphology
The surface morphology of the keratin films before and after crosslinking and heparin immobilization was evaluated by SEM, and the representative images are shown in Figure 12. Surface morphology of keratin films observed by SEM. SEM images of untreated keratin films, EDC-crosslinked keratin films, and heparin/AEM-immobilized keratin films. No substantial morphological differences were observed after crosslinking or heparin immobilization under the present observation conditions.
The untreated keratin film exhibited a relatively smooth and continuous surface without apparent defects or phase separation. After crosslinking, no significant changes in surface morphology were observed, and the film maintained a similar smooth and homogeneous appearance. Likewise, the heparin/AEM-immobilized film showed no noticeable alteration in surface features compared to the untreated and crosslinked samples.
These results indicate that both crosslinking and subsequent heparin immobilization did not induce substantial morphological disruption or surface roughening of the keratin films, suggesting that the chemical modifications proceeded without compromising the structural integrity of the material.
On the other hand, slight differences in apparent film thickness near the sample edges were observed, where the crosslinked and heparin-immobilized samples appeared marginally thicker than the untreated film. This difference is attributed to film contraction during the aqueous treatment and subsequent drying processes, leading to densification of the film structure.
Overall, the SEM observations confirm that the applied surface modification strategy preserves the macroscopic and microscopic morphology of the keratin films while inducing only minor structural rearrangements associated with processing conditions.
Surface wettability
The surface wettability of keratin films before and after surface modification was evaluated by static water contact angle measurements (Figure 13). The untreated keratin films exhibited a contact angle of approximately 92°, indicating a moderately hydrophobic surface. After EDC crosslinking, the contact angle slightly increased to around 100°, although no statistically significant difference was observed, suggesting a minor reduction in surface hydrophilicity due to the consumption of polar functional groups. Water contact angles of keratin films before and after surface modification. Comparison of static water contact angles for untreated, EDC-crosslinked, and heparin/AEM-immobilized keratin films. Heparin immobilization significantly decreased the contact angle, indicating increased surface hydrophilicity. Data are presented as mean ± SD (n = 6). Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparison test. ns: not significant; *p < 0.05; **p < 0.01.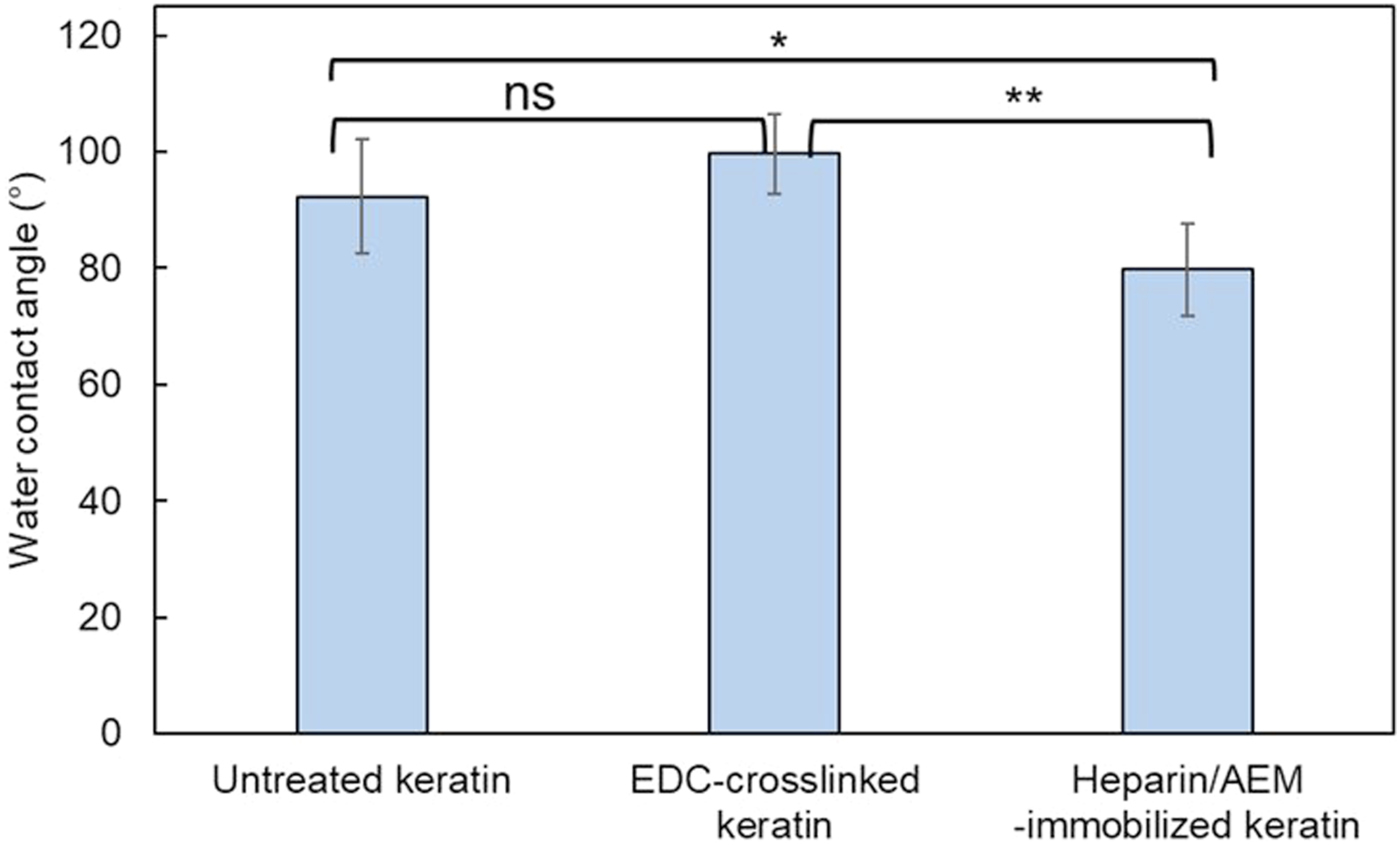
In contrast, the heparin/AEM-immobilized keratin films showed a significantly lower contact angle of approximately 80°, indicating an increase in surface hydrophilicity compared to the EDC-crosslinked films. This decrease can be attributed to the introduction of hydrophilic functional groups derived from heparin, such as sulfate (–SO3−) and hydroxyl (–OH) groups, which enhance the affinity for water at the material surface.
Notably, the reduction in contact angle was moderate rather than drastic, which is consistent with the relatively low surface coverage of immobilized heparin and the dominant contribution of the keratin substrate. Nevertheless, the statistically significant decrease supports successful surface functionalization. Furthermore, the covalent immobilization of heparin via thiol–maleimide coupling is expected to provide stable surface presentation of these hydrophilic moieties, in contrast to physically adsorbed heparin, which may desorb during washing.
From a biomaterials perspective, even a moderate increase in surface hydrophilicity can contribute to improved hemocompatibility by reducing nonspecific protein adsorption. Hydrophilic surfaces are known to suppress fibrinogen adsorption and subsequent platelet adhesion, which are critical steps in thrombus formation. Therefore, the present results are consistent with the anticoagulant behavior .
Overall, these results demonstrate that thiol–maleimide coupling effectively modulates the surface properties of keratin films. This combined effect of biochemical functionality and physicochemical surface modification plays an important role in the overall anticoagulant performance of the material.
Mechanical properties
The mechanical properties of keratin films were evaluated by tensile testing, and the results are summarized in Figure 14. The apparent Young’s modulus and tensile strength are shown in Figure 14(a) and (b), respectively. Mechanical properties of keratin films before and after heparin immobilization. (a) Apparent Young’s modulus. (b) Tensile strength. Data are presented as mean ± SD. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparison test. *p < 0.05 versus EDC-crosslinked keratin films.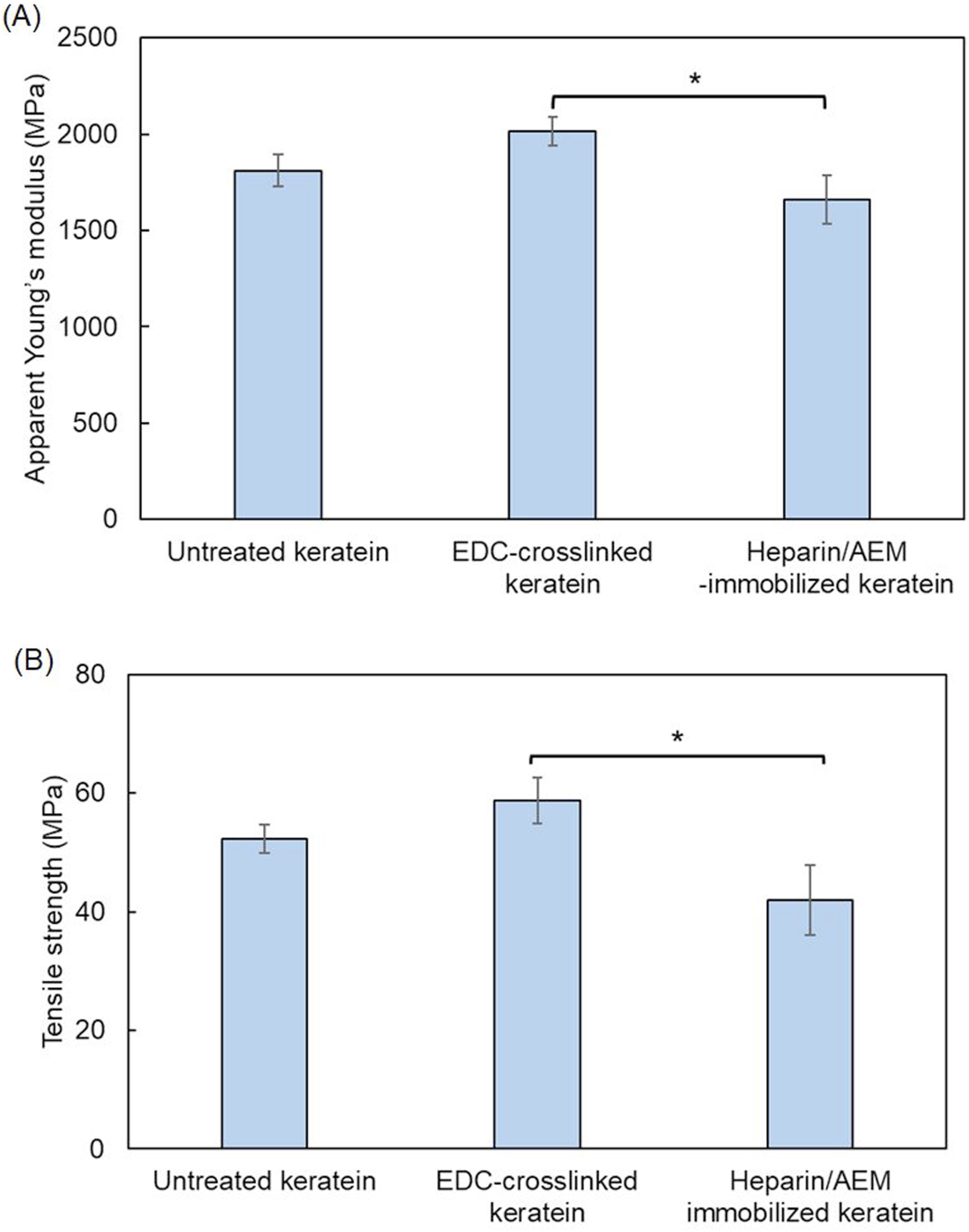
As shown in Figure 14(a), the apparent Young’s modulus of the EDC-crosslinked keratin films was higher than that of the untreated films, indicating that crosslinking effectively increased the stiffness of the keratin network. This increase can be attributed to the formation of additional intermolecular bonds, which restrict molecular mobility and enhance resistance to deformation.
In contrast, the heparin/AEM-immobilized keratin films exhibited a decrease in apparent Young’s modulus compared with the crosslinked films. A statistically significant difference was observed between the crosslinked and heparin-functionalized groups (*p < 0.05). This reduction in stiffness is likely associated with structural changes induced during the immersion and redrying processes, as well as increased hydrophilicity following heparin immobilization, which may alter intermolecular interactions within the film.
A similar trend was observed for tensile strength (Figure 14(b)). The EDC-crosslinked keratin films showed higher tensile strength than the untreated films, suggesting improved mechanical integrity due to network formation. In contrast, the heparin/AEM-immobilized films exhibited a significant decrease in tensile strength compared with the crosslinked films (*p < 0.05). This decrease may be attributed to partial disruption or reorganization of the network structure during the post-modification process, as well as increased structural heterogeneity.
Overall, these results indicate that crosslinking enhances both stiffness and strength of keratin films, whereas subsequent heparin immobilization leads to a moderate reduction in mechanical properties. This behavior reflects a trade-off between mechanical reinforcement and functional surface modification.
Evaluation of anticoagulant activity of heparin-immobilized keratin
The anticoagulant response of heparin-immobilized keratin films was evaluated by comparing thrombin-induced coagulation behavior with various control materials, as shown in Figure 15. Unmodified keratin films exhibited coagulation behavior comparable to that of the thrombin-only control, indicating that native keratin does not exhibit intrinsic anticoagulant activity and does not measurably interfere with the coagulation process under the tested conditions. Anticoagulant activity of heparin-immobilized keratin films. Relative coagulation rates calculated from the initial slope of the absorbance–time curves for thrombin-only control, untreated keratin films, heparin-immobilized keratin films, and Pellethane®. Data are presented as mean ± SD. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparison test. ***p < 0.001 versus heparin-immobilized keratin films.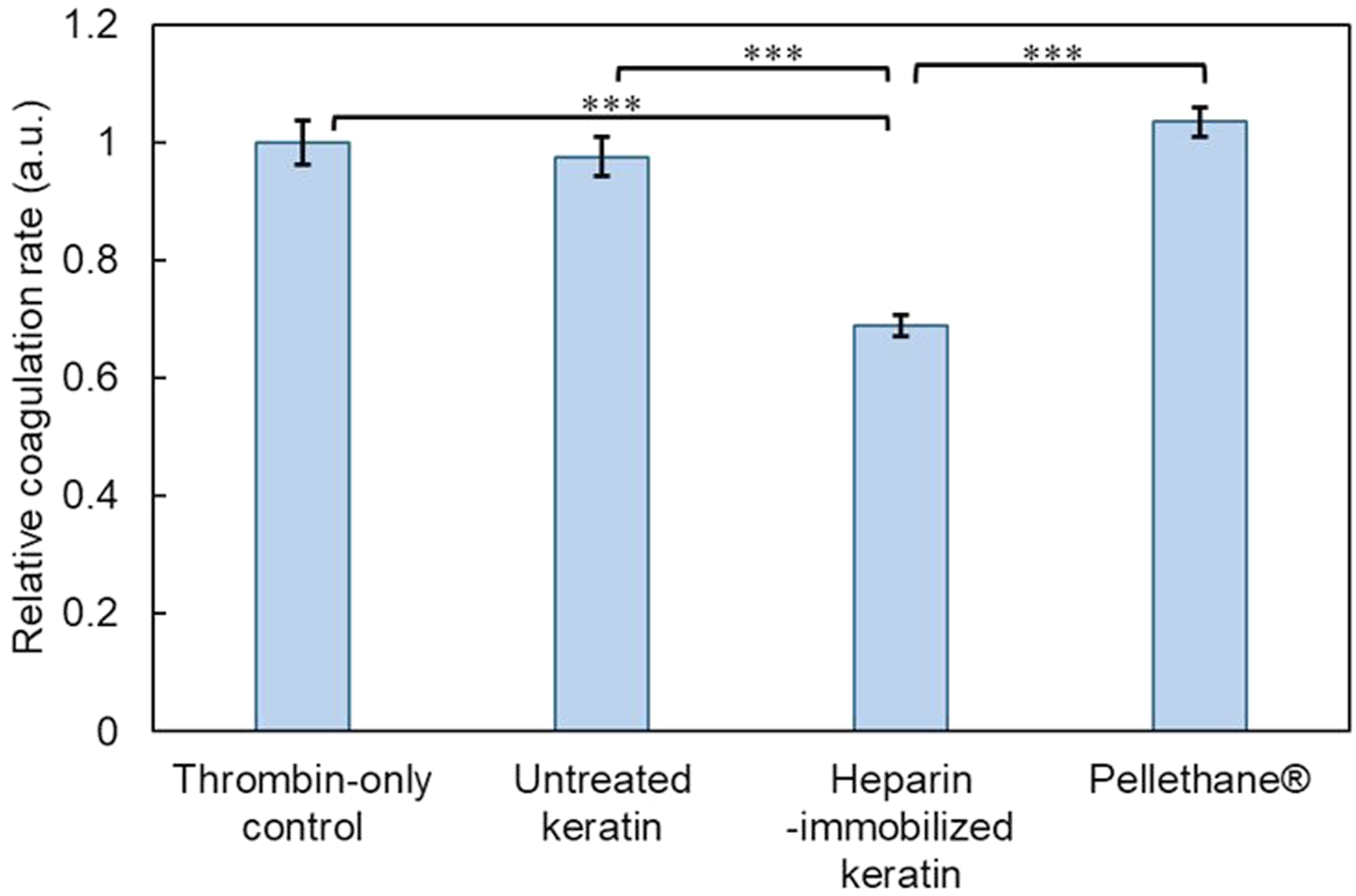
In contrast, heparin-immobilized keratin films significantly suppressed fibrin formation, indicating that immobilized heparin retained thrombin-inhibitory functionality after covalent attachment. The relative coagulation rate, calculated from the initial slope of the absorbance–time curve at 405 nm, was significantly lower for the heparin-immobilized keratin films than for the untreated keratin and Pellethane® control groups. These findings demonstrate that surface-immobilized heparin remained functionally active even after covalent conjugation onto the keratin substrate.
In addition, clotting behavior estimated from the absorbance–time profiles obtained in the plasma clotting assay (Figure 6) showed rapid clot formation for the thrombin-only control, whereas free heparin and the heparin/AEM complex strongly suppressed fibrin formation within the measurement period. Compared with free heparin in solution, the anticoagulant response of the immobilized system was relatively moderate, which is consistent with the restricted molecular accessibility of immobilized heparin at a heterogeneous solid–liquid interface.
These observations are in agreement with previous studies reporting anticoagulant responses in keratin-based or sulfated biomaterial surfaces, supporting the applicability of thiol-mediated immobilization strategies.23,46 Importantly, the preservation of anticoagulant functionality in the immobilized state, together with the absence of detectable heparin release (Figure 9) and the surface chemical evidence obtained by XPS and FT-IR analyses (Figure 11), highlights the potential utility of this approach for surface modification of blood-contacting biomaterials. In contrast, Pellethane®—a clinically used polyurethane—did not exhibit anticoagulant activity under identical conditions, serving as a reference material. 47
Overall, these results demonstrate that thiol-mediated heparin immobilization on keratin films enables stable anticoagulant surface functionality without relying on physically adsorbed anticoagulants. This strategy provides a sustainable and animal-free platform for anticoagulant surface modification of protein-based biomaterials.
Conclusions
This study demonstrates that human hair–derived keratin represents a safe, sustainable, and functionally versatile protein resource for biomaterial design. By employing a reductive extraction system combining guanidine hydrochloride and tris(hydroxypropyl)phosphine (THPP), keratin was obtained with a high extraction efficiency while preserving reactive thiol (–SH) functionality.
The retained thiol groups provided chemically selective reaction sites for subsequent biofunctionalization under mild aqueous conditions. Using maleimide–thiol coupling chemistry, heparin—a clinically established anticoagulant—was covalently immobilized onto keratin films through an aminoethyl maleimide (AEM) linker. Functional evaluation using thrombin-induced clotting assays indicated that immobilized heparin retained anticoagulant activity, whereas unmodified keratin films did not measurably affect coagulation behavior under the tested conditions.
These results highlight the utility of keratin’s native thiol groups as reactive handles for covalent attachment of bioactive molecules, expanding the chemical versatility of protein-based biomaterials beyond conventional amino- or carboxyl-directed modifications. Importantly, the covalent immobilization strategy enabled stable presentation of heparin without detectable leaching, supporting the robustness of the thiol-mediated conjugation approach.
Overall, this work establishes a thiol-mediated molecular engineering platform for anticoagulant surface modification using animal-free keratin-based materials. The developed keratin–heparin films provide a chemically stable anticoagulant surface response, suggesting their potential applicability as coatings or interface materials for blood-contacting biomedical devices. Future studies may extend this platform toward immobilization of other functional biomolecules, enabling broader exploration of keratin-based systems for advanced biomaterial applications.48–52
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.