
Abstract
Hot–pressed nylon–66 is widely used in engineering applications, and density functional theory (DFT) calculations and molecular dynamics (MD) simulations played the positive roles in the improvements of mechanical and tribological properties. In this study, the influences of hot–pressing process on the crystalline structure and intermolecular interactions were investigated through the combination of DFT calculations and MD simulations. The results demonstrate that the hot–pressing process enhances the molecular chain alignment and enhances intermolecular interactions, which leads to the increase of mechanical properties with the tensile strength of X% and the decrease of coefficient of friction (COF) by Y%) and the reduction of wear rate. This offers the valuable insights into the structure–property relationships of hot–pressed nylon–66, providing the theoretical guidance for the design of high–performance polymer tribo–materials. The findings suggest that this material has the great potential for applications in environments requiring high mechanical strength and low friction.
Keywords
Introduction
Polyhexamethylene adipamide (nylon–66, (C12H22N2O2)n) is one of the most widely used engineering thermoplastics due to its excellent mechanical strength, thermal stability, chemical resistance and self–lubricating properties, making it suitable for applications in automotive components, gears, bearings and other tribological parts subjected to dry or boundary lubrication conditions. 1 The friction and wear behavior of nylon–66 are the critical factors determining the service life, reliability and energy efficiency of mechanical system. 2 Consequently, improving the mechanical and tribological properties of nylon–66 remains the essential area of research in the polymer tribo–materials. In practical applications, the friction and wear behavior of nylon–66 directly determines the service life, reliability and energy efficiency of mechanical systems. 3 Therefore, improving the mechanical and tribological properties of nylon–66 remains an important research topic in polymer tribo–materials.
At present, the processing methods play a critical role in determining the microstructure and macroscopic properties of nylon–66. 4 Among various fabrication techniques, hot–pressing has attracted increasing attention due to its ability to promote the molecular chain rearrangement, enhance the crystallinity and improve the interfacial bonding without the use of additives or reinforcements. 5 Compared with conventional injection molding or extrusion, the hot–pressed nylon–66 exhibits the superior density, mechanical strength and wear resistance. 6 However, the underlying mechanisms of hot–pressing altering the microstructure affects the mechanical and tribological behaviors are not yet completely understood, 7 particularly from the atomistic and molecular perspectives.
Previous studies have reported that the tribological properties of nylon–66 is closely related to its crystalline structure, molecular orientation and intermolecular interactions, especially hydrogen bonding between the amide groups. 8 Nevertheless, the conventional experimental techniques are limited in revealing the intrinsic mechanisms at the electronic and atomic scales, and the computational approaches, such as density functional theory (DFT) and molecular dynamics (MD) simulations provide the powerful tools to bridge this gap.9–11 Among them, the DFT calculations can offer the fundamental insights into electronic structure, binding energy and hydrogen bonding characteristics 12 ; while the MD simulations enable the investigation of mechanical response, deformation behavior and frictional processes under the realistic loading and sliding conditions. 13 Despite the growing use of multiscale simulations on the polymer research, the systematical investigation of hot–pressed nylon–66combination through the experimental characterization with the DFT and MD simulations are still scarce. 14 In particular, the relationship between the hot–pressing–induced microstructural evolution and tribological properties at the atomic and molecular scales have not been comprehensively clarified, and most existing works focus either on macroscopic tribological testing or on molecular simulations of idealized nylon–66 models, lacking the direct correlation between the processing, structure and performance. 15
In this study, the microstructure, mechanical properties and tribological behavior of hot–pressed nylon–66 were investigated through the experimental and computational approaches, and the effects of hot–pressing on the molecular chain alignment and intermolecular interactions were also discussed, where the DFT calculations elucidated the electronic structure and hydrogen bonding at the atomic scale, while the MD simulations explored the mechanical and tribological properties. By integrating experimental results with the multiscale simulations, the aim to clarify the structure–property–tribology relationships of hot–pressed nylon–66, offering the new insights for the design of high–performance polymer tribo–materials.
Experimental details
Sample Preparations
The commercial nylon–66 pellets, dried in a vacuum oven at 80–100°C for 12 h to remove the moisture and prevent the hydrolysis, were used as the raw material for the hot–pressing process. The testing samples were fabricated using a hot–pressing machine, and the nylon–66 pellets were placed into a stainless–steel mold. The hot–pressing was carried out at 180°C, and the hot–pressing time was set as 5, 10 and 20 min to investigate the effect of hot–pressing time on the microstructure and properties of nylon–66. After the designated holding time, the samples were cooled to room temperature, and the samples required for the experiment were obtained.
Microstructural characterizations
The morphologies and elemental distributions of hot–pressed nylon–66 samples were analyzed using a QUANTA FEG 450 type field emission scanning electron microscope (FE–SEM) and its configured energy dispersive spectroscope (EDS), respectively, and the chemical states of analyzed using an Escalab 250Xi type X–ray photoelectron spectrometer (XPS). The Fourier transform infrared (FTIR) spectrum was conducted on a Nicolet–iS10 type spectrometer in the measurement range of 500–3600 cm−1.
Mechanical and tribological tests
The mechanical properties of hot–pressed nylon–66 samples were evaluated using an UTM2102 type electronic universal machine at the constant crosshead speed of 10 mm min−1, following the ASTM standard. Each test was repeated three times to ensure the reproducibility, and the average value was presented as the experimental result.
The tribological properties were investigated using a TriboStudio TS20 type wear ball–on–disk tribometer under the dry sliding condition, and the Si3N4 ball with the diameter of 4 mm was used as the tribo–pair. The normal load, sliding speed and operation time were set to 10 N, 500 rpm, and 30 min, respectively, and the coefficient of friction (COF) was continuously recorded during the tribological test.
DFT calculations and MD simulations
The DFT calculations and MD simulations were employed to analyze the wear mechanisms of hot–pressed nylon–66, which were used to explore its microscopic deformation, molecular interactions and structural evolution during the wear process. In this case, the specific focus on how the hot–pressing time influenced the molecular packing, hydrogen bonding, mechanical properties and wear resistance. Among them, the DFT calculations were performed to gain the deeper understanding of electronic structure, intermolecular interactions and intrinsic strengthening mechanisms in the nylon–66, and its molecular models of nylon–66 were constructed using the generalized gradient approximation (GGA) with the perdew–burke–ernzerhof (PBE) functional. The plane–wave basis set with an energy cutoff of 400 eV was adopted, and the Van der Waals interactions were explicitly considered to accurately capture the intermolecular forces. By analyzing the binding energy and hydrogen bonding characteristics of nylon–66 under the different chain packing states, and how variations of molecular alignment and packing influence the structure and properties was revealed. The effects of hot–pressing times on the chain packing and structural evolution were detailed, offering the insights into the microscopic changes that influenced the macroscopic properties.
For the MD simulations, the multiscale study was we conducted to analyze the mechanical properties and tribological behavior of hot–pressed nylon–66, and the models of nylon–66 were constructed with the varying molecular orientations and chain packing densities to represent the effects of hot–pressing conditions on the material. The models were equilibrated under constant temperature and pressure conditions using the NVT ensemble (constant N, V and T), which was suitable for the simulating materials under realistic processing conditions. The force field was adopted to describe the atomic interactions, and the periodic boundary conditions were applied in all directions to simulate the bulk behavior. In this case, the time step was set to 1 fs to ensure simulation accuracy.
Moreover, the different hot–pressing time was represented by varying the chain packing density and molecular orientation in the nylon–66 models, and the mechanical properties were evaluated by the uniaxial tensile simulations; while the tribological behavior was investigated using the sliding model under the constant normal load and sliding speed, and the COF was obtained from the steady–state tangential force during the sliding–wear process.
Results and discussion
SEM images and mapping analysis
Understanding the morphological evolution of nylon–66 samples during the hot–pressing process was crucial for correlating the processing conditions with the material properties, and their structure of chain alignment at the different hot–pressing time directly affected their mechanical and tribological properties.
16
Figure 1 presents the SEM and mapping images of nylon–66 particles and hot–pressed samples. The surface of nylon–66 particle exhibited the irregular folds and pores (Figure 1(a)), indicating the relatively loose packing of polymer chains.
17
After the hot–pressing for 5 min, the surface of nylon–66 sample became more uniform and compact, and the lamellar structure began to align (Figure 1(b)), suggesting that the partial chain orientation was induced by the pressure and heat effect.18,19 As the hot–pressing time was prolonged to 10 min, the densification and alignment of polymer chains were enhanced (Figure 1(c)), as evidenced by the smoother surface and more continuous lamellar feature. For the 20-min hot–pressed sample, the surface of nylon–66 sample appeared highly uniform and densely packed with the minimal voids (Figure 1(d)), indicating that the extended hot–pressing time promoted the near–complete chain rearrangement and crystallite alignment.
20
SEM images of nylon–66 particle (a) and hot–pressed nylon–66 samples at 5 (b), 10 (c) and 20 (d) min.
The elemental mapping confirmed the uniform distributions of C, O, N and S across all samples, which were consistent with the chemical composition of nylon–66. Notably, as the hot–pressing time increased, the intensity of N and O appeared more homogeneous, reflecting the improved packing and reduced the surface defects. 4 These observations suggested that the longer hot–pressing time enhanced the molecular chain orientation and intermolecular interactions, which contributed to the improved mechanical and tribological properties in the subsequent tests.
FTIR and XPS analyses
To elucidate the influence of hot–pressing time on the molecular structure and surface chemistry of hot–pressed nylon–66 samples, it was essential to investigate how the hot–pressing time affected the intermolecular interactions and surface functional groups, which helped to understand their molecular–level structures.
21
Figure 2 presents the FTIR and XPS analyses of hot–pressed nylon–66 samples at the different hot–pressing time, providing the insight into the evolution of chemical structure and surface composition induced by the hot–pressing time. All samples exhibited the characteristic FTIR absorption bands of nylon–66 (Figure 2(a)), confirming that the hot–pressing time did not alter the fundamental chemical structure.
22
The absorption bands at ∼3300 cm−1 (N–H stretching), ∼2930 and ∼2860 cm−1 (C–H stretching), ∼1630 cm−1 (C = O stretching, amide I) and ∼1540 cm−1 (N–H bending and C–N stretching, amide II), as well as those around ∼1200–1300 cm−1 (C–N stretching),
23
were clearly observed. The intensities of amide–related peaks, particularly the C = O and N–H vibrations, showed the gradual enhancement, indicating the strengthened intermolecular hydrogen bonding and improved chain packing.
24
FTIR spectra (500–2000 cm−1) of hot–pressed nylon–66 samples (a), FTIR (2500–3500 cm−1) of hot–pressed nylon–66 samples (b), XPS survey spectra of hot–pressed nylon–66 samples (c), C1s spectra (d), O1s spectra (e) and N1s (f).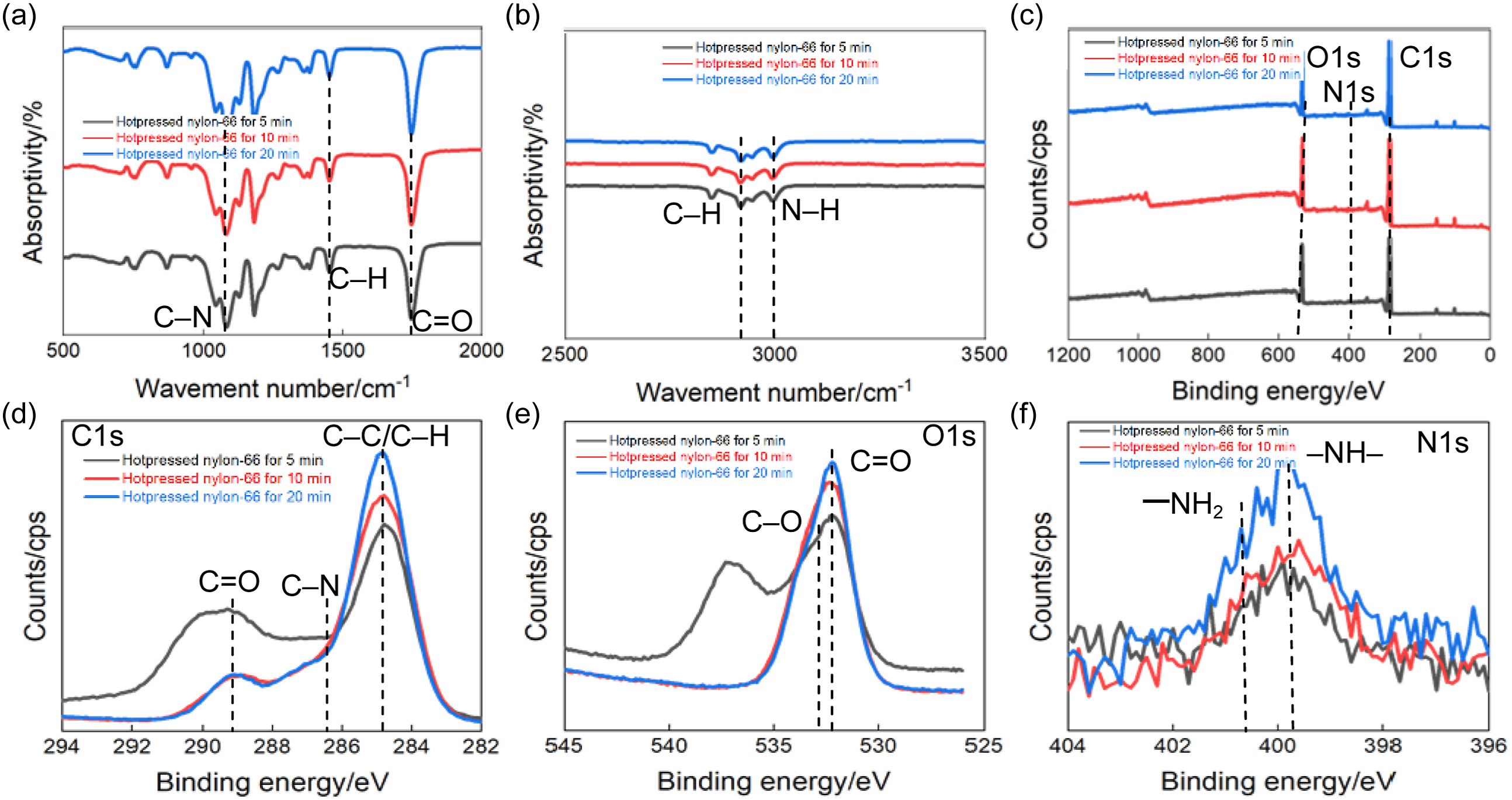
The enlarged FTIR spectra in the range of 2500–3500 cm−1 (Figure 2(b)) further indicated the evolution of hydrogen–bond–related functional groups. The N–H stretching band became slightly broader and more obvious for the longer–time hot–pressed samples, suggesting the increased hydrogen–bond density due to the enhanced molecular mobility and rearrangement during the prolonged pressing time. This behavior was favorable to improving the mechanical and tribological properties of nylon–66 samples at the longer hot–pressing time.
Figure 2(c) shows the XPS survey spectra of hot–pressed nylon–66 samples, where the characteristic peaks of C1s, O1s and N1s were clearly identified, confirming the presences of C, O and N on all samples. Notably, the intensities of O1s and N1s peaks were increased with the hot–pressing time, indicating the higher content of polar functional groups on the surface, which was related to the chain reorientation and surface reconstruction during the hot–pressing process. 25
The high–resolution XPS spectra provided the further details on the chemical states of surface elements. The C1s spectra were deconvoluted into C–C/C–H (∼284.8 eV), C–N (∼286.0 eV), and C = O (∼288.0 eV) peaks (Figure 2(d)), and the contributions of C–N and C = O became more prominent as the hot–pressing time increased, suggesting the increased proportion of amide linkages participating in the intermolecular interactions. 26 In Figure 2(e), the O1s spectra exhibited the two main components attributed to C–O and C = O bonds, and the C = O peak intensity increased for the longer hot–pressing time, further confirming the enhanced amide bonding. 27 Similarly, the N1s spectra (Figure 2(f)) was assigned to –NH– and –NH2 species, and their increased intensity and slight peak sharpening for the 20-min hot–pressed sample indicated the more uniform chemical environment and stronger hydrogen–bond interactions om its surface. 28 As a result, the combined FTIR and XPS results demonstrated that the extension of hot–pressing time did not change the chemical composition of nylon–66, but significantly enhanced the intermolecular interactions, particularly hydrogen bonding and chain packing. These chemical and surface structural evolutions provided the strong molecular–level insight into the improvements in the mechanical strength and tribological stability of nylon–66 sample as the hot–pressing time increased, which facilitated the increased molecular mobility and better alignment of polymer chains.
DFT calculation and MD models
To elucidate the microscopic structural evolution and thermal response of nylon–66 samples during the hot–pressing process, the DFT calculations and MD simulations were systematically employed to analyze the atomic interactions and molecular behaviors. These simulations aimed to reveal the relationship among the molecular configuration, temperature and hot–pressing time, providing the fundamental insights into the processing–structure–performance correlations of hot–pressed nylon–66 samples.
29
Figure 3 displays the optimized molecular structure and the dynamic evolution of hot–pressed nylon–66 samples under the different hot–pressing conditions at the multiple length and time scales. The optimized molecular configuration of hot–pressed nylon–66 samples obtained from the DFT calculations (Figure 3(a)) exhibited the relatively linear backbone with the periodically distributed amide groups.
30
The distinct charge accumulation and depletion regions were observed around the amide functionalities, indicating the strong polarity along the polymer chain. These electronic characteristics were favorable for the formation of intermolecular hydrogen bonding, which played a crucial role in determining the structural stability and intermolecular interactions of hot–pressed nylon–66 samples. DFT calculations of hot–pressed nylon–66 samples (a). MD models of hot–pressed nylon–66 samples at different temperatures (b) and MD models of nylon–66 at different hot–pressing time (c).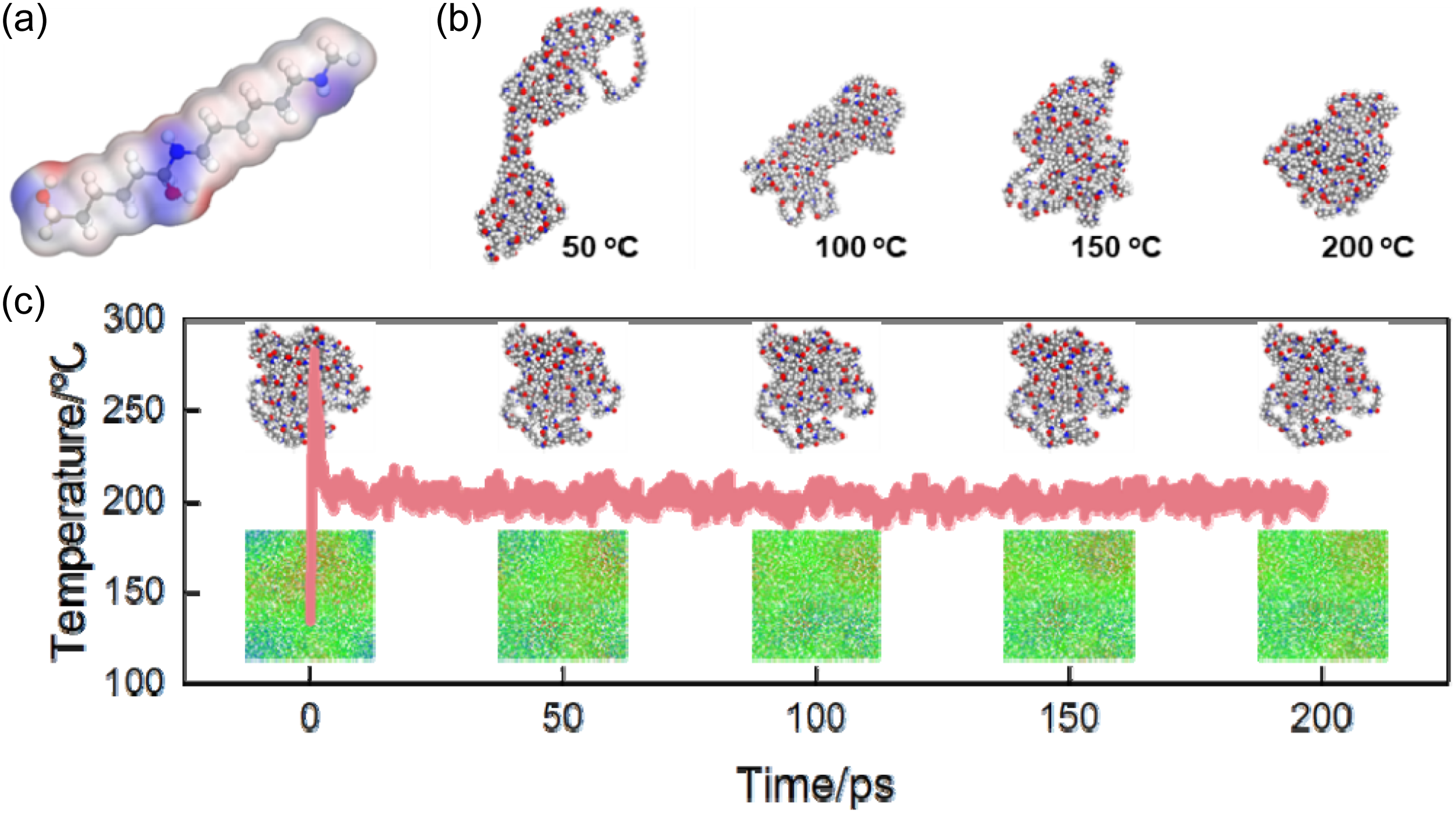
Figure 3(b) presents the MD simulation snapshots of hot–pressed nylon–66 samples at the different temperatures (50, 100, 150 and 200°C). At the lower temperatures, the polymer chains displayed the relatively disordered and loosely packed conformations with the chain bending. As the temperature increased, the thermal motion of molecular chains became more intense, enabling the system to overcome the conformational energy barriers. Consequently, the polymer chains were gradually rearranged into more compact and homogeneous structures. At the higher temperatures (150–200°C), the molecular aggregation became significantly denser, suggesting that the elevated temperature facilitated the chain mobility and promoted the formation of thermodynamically more stable configuration. 31
Figure 3(c) further depicts the molecular structures of hot–pressed nylon–66 samples versus simulation time evolutions during the hot–pressing process. The temperature profiles indicated the rapid increase at the initial stage, followed by stabilization at ∼200°C. The corresponding MD snapshots revealed that the polymer chains initially exhibited the relatively random distribution. As the hot–pressing time increased, the chains underwent the continuous rearrangement and densification, accompanied by the noticeable reduction of free volume and voids in the system. 32 This structural evolution demonstrated that the extended hot–pressing time enhanced the molecular diffusion and chain packing efficiency, leading to the more uniform and compact microstructure.
Overall, the combined DFT and MD results indicated that the temperature and hot–pressing time were the critical factors governing the conformational evolution and structural densification of hot–pressed nylon–66 samples, and the increased temperature and extended hot–pressing time effectively promoted the chain mobility, intermolecular interactions and structural stabilization, which provided the fundamental insights into the processing–structure relationship of hot–pressed nylon–66 samples and macroscopic thermomechanical performance.
Mechanical properties
To further clarify the influence of hot–pressing time on the macroscopic mechanical behavior of hot–pressed nylon–66 samples, the uniaxial tensile tests were conducted on the samples fabricated at the different hot–pressing time. The tensile stress–strain responses and digital image correlation (DIC) strain field analysis were employed to quantitatively evaluate the effects of hot–pressing time on the strength, stiffness, ductility and deformation homogeneity of hot–pressed nylon–66 samples.
33
Figure 4 presents the tensile stress–strain responses of hot–pressed nylon–66 samples at the different hot–pressing time, demonstrating the strong dependence of mechanical properties on the hot–pressing time. For the 5-min hot–pressed nylon–66 sample, the stress increased with the strain and reached the relatively low maximal value of 60–70 MPa before the premature failure at the strain of 2.2–2.4%. This behavior indicated that the insufficient consolidation was associated with the incomplete polymer chain diffusion and the presence of residual pores or weak interfacial bonding, which limited the effective stress transfer under the tensile loading action.
34
Mechanical testing results of hot–pressed nylon–66 samples at different hot–pressing time.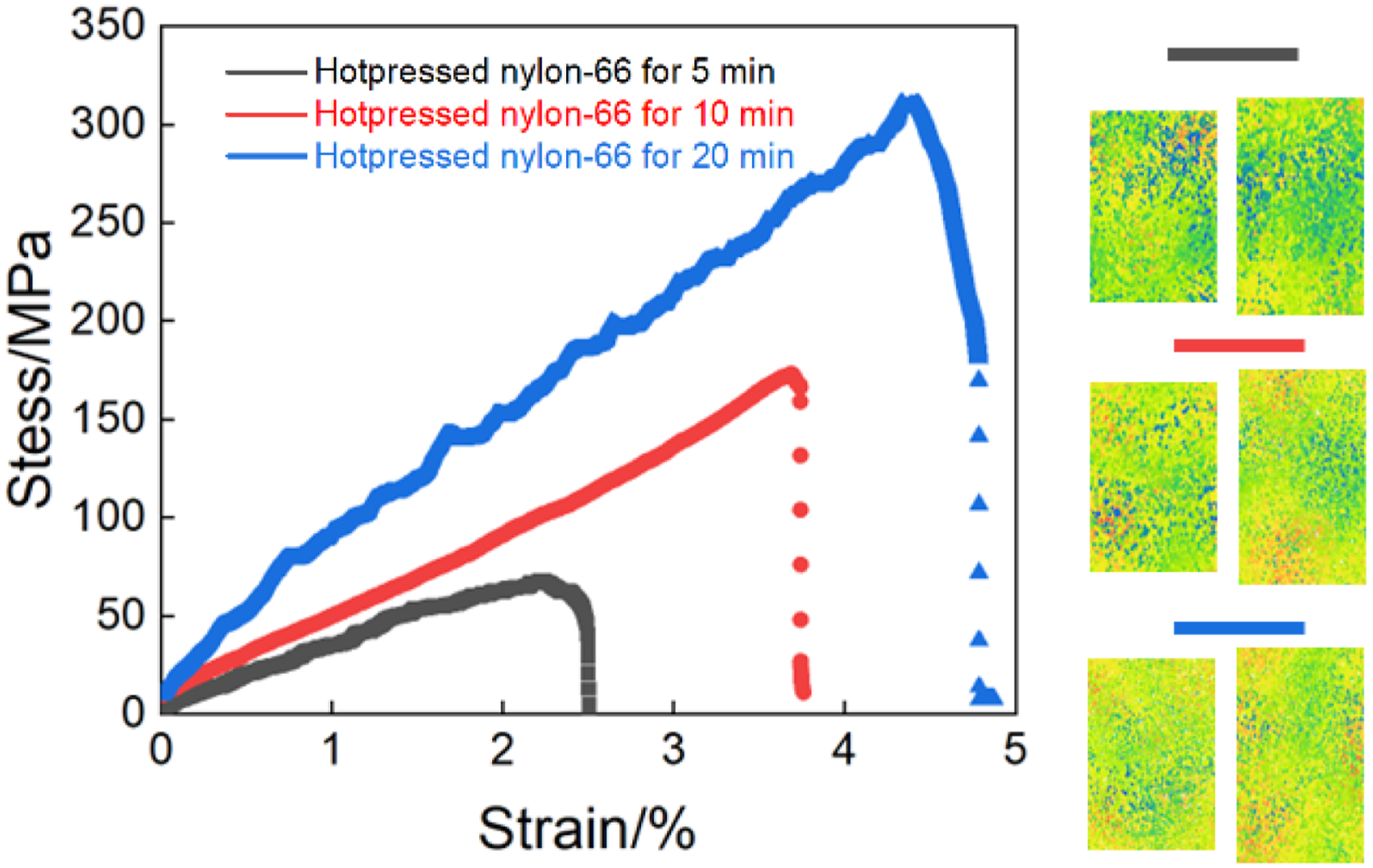
As the hot–pressing time was increased to 10 min, the obvious enhancement of mechanical properties was observed. The slope of initial linear region became steeper, reflecting the increase of elastic modulus; while the ultimate tensile strength increased significantly to 160–170 MPa. Simultaneously, the fracture strain increased to 3.5–4.0%, suggesting the improved ductility. This improvement was attributed to more effective melting and rearrangement of polymer chains during the hot–pressing process, resulting in the higher densification, reduced defect content and stronger intermolecular interactions. 35
The 20-min hot–pressed nylon–66 sample exhibited the best overall mechanical properties, and its stress–strain curve showed the nearly linear stress increase over the wide strain range, followed by the high peak stress exceeding 300 MPa, and the fracture strain approached 5%. The combination of high strength and large elongation indicated the well–consolidated microstructure with the efficient load transfer, which enhanced the resistance to the crack initiation and propagation. The gradual stress dropped after the peak also implied the more progressive damage evolution rather than abrupt brittle failure. 36
The DIC strain maps on the right further supported these observations, and the 5-min hot–pressed sample displayed the highly localized strain concentrations prior to failure, revealing the presence of structural heterogeneities. In contrast, the 10-min and 20-min hot–pressed nylon–66 samples showed more homogeneous strain distributions across the gauge section, indicating that the uniform deformation delayed the strain localization. Overall, the extension of hot–pressing time effectively promoted the structural densification and microstructural uniformity of hot–pressed nylon–66 samples, leading to the substantial improvements of tensile strength, stiffness and ductility.
Tribological properties
To understand the effect of hot–pressing time on the tribological performance of hot–pressed nylon–66 samples, it was crucial to investigate how the hot–pressing time influenced the macroscopic tribological behavior and microscopic wear mechanisms. The hot–pressing time affected the densification, molecular orientation and intermolecular interactions of polymer matrix, which in turn determined its response under the sliding–wear contact.
37
Figure 5 illustrates the tribological behavior and corresponding wear mechanisms of hot–pressed nylon–66 samples at the different hot–pressing time. The COFs of all samples were increased with the sliding time (Figure 5(a)), indicating the typical running–in (RI) process followed by the quasi–steady friction state. The 5-min hot–pressed nylon–66 sample exhibited the lowest initial COF, but also showed the obvious fluctuations and faster increase during the sliding–wear process, suggesting the unstable interfacial contact and poor wear resistance. In contrast, the 10-min hot–pressed nylon–66 sample displayed the more moderate increase of COF and reduced fluctuation amplitude, implying the improved surface integrity and load–bearing capacity. The 20-min hot–pressed nylon–66 sample presented the highest COF throughout the tribological test; however, its COF curve was comparatively smoother, indicating the more stable frictional interface at the extended hot–pressing time. This behavior was attributed to the higher degree of densification and stronger intermolecular interactions during the extending hot–pressing process, which enhanced the resistance to surface deformation and material removal.
38
COFs versus sliding time curves (a), system energy of hot–pressed nylon–66 samples at different hot–pressing time (b) and wear models of 5– (c), 10– (d) and 20– (e) min hot–pressed nylon–66 samples.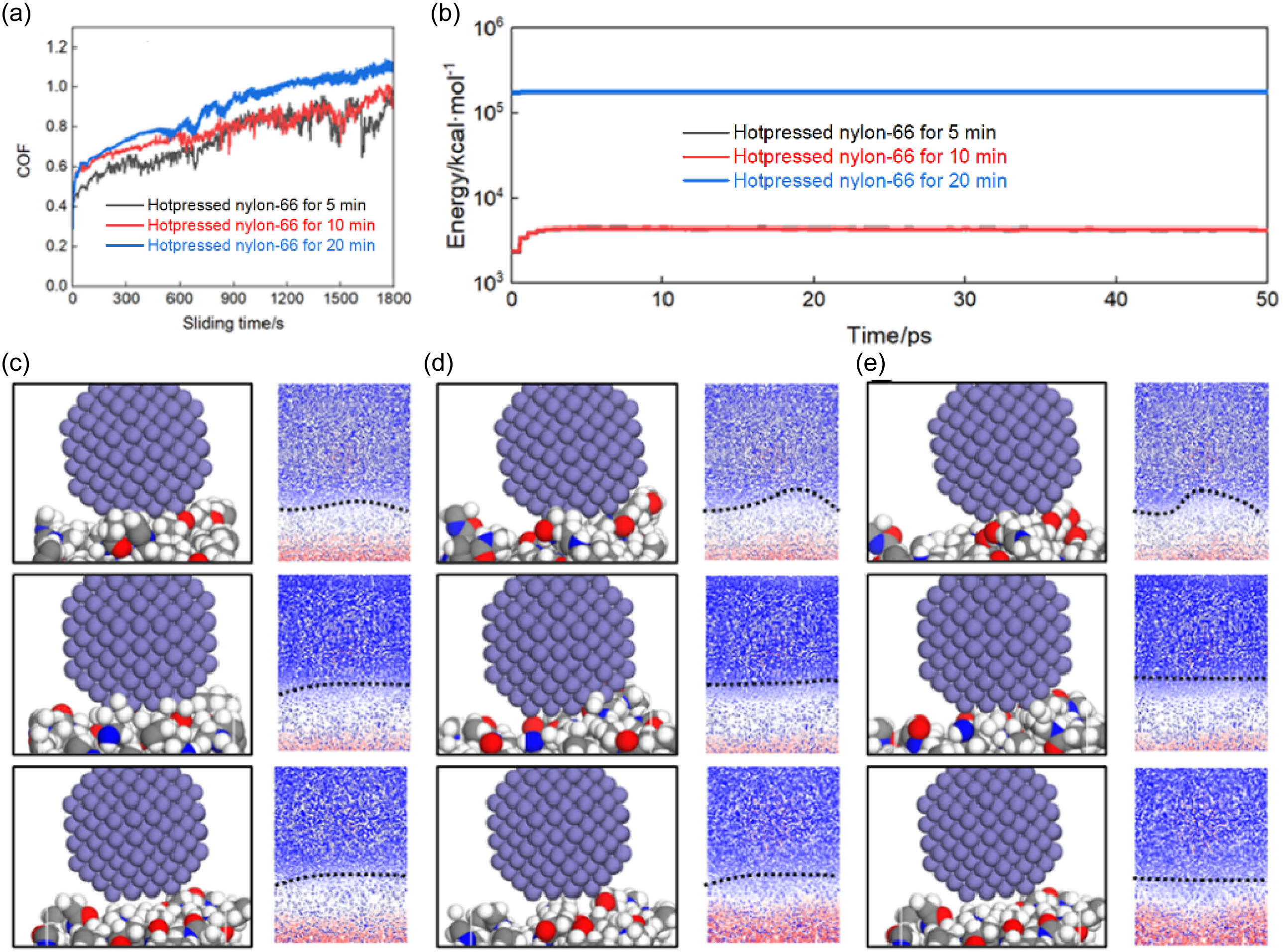
Figure 5(b) shows the evolution of total system energy obtained from the MD simulations for the hot–pressed nylon–66 samples at the different hot–pressing time. The system energy remained nearly constant during the sliding–wear process for all cases, indicating that the simulated systems quickly reached the dynamic equilibrium. Notably, the absolute energy level was increased with the hot–pressing time, and the 20-min hot–pressed nylon–66 sample exhibited the highest system energy, followed by the 10– and 5-min hot–pressed nylon–66 samples. This trend reflected the increased cohesive energy density and structural stability of hot–pressed nylon–66 samples after the longer hot–pressing time, which required the higher energy to deform and disrupt at the sliding interface. 39
The atomistic wear models in Figure 5(c)–(e) further revealed the underlying wear mechanisms at the different hot–pressing time. For the 5-min hot–pressed nylon–66 samples, the severe interfacial disorder and significant polymer chain displacement were observed beneath the tribo–pair, accompanied by the obvious material accumulation and local densification on the near–surface region (Figure 5(c)). This indicated the adhesive–dominated wear and extensive plastic deformation due to the weak intermolecular bonding. 40 As the hot–pressing time increased to 10 min, the subsurface deformation became less severe, and the polymer chains showed more coordinated motion (Figure 5(d)), suggesting the improved load transfer and reduced adhesive wear. For the 20-min hot–pressed nylon–66 sample, the polymer matrix maintained the more ordered structure with the minimal chain pulling–out and relatively stable subsurface deformation zone (Figure 5(e)). The corresponding density distributions showed the smoother gradient across the interface, indicating the suppressed wear and enhanced structural integrity. 41
Overall, the increase of hot–pressing time improved the tribological stability and wear resistance of hot–pressed nylon–66 samples. Although the higher COF was observed for the extensively hot–pressed nylon–66 sample, the frictional process became more stable, and the wear mechanism was transferred from severe adhesive wear to more controlled and mild deformation–dominated state, consistent with the enhanced mechanical properties and densified microstructure induced by the prolonged hot–pressing time.
In these cases, the MD simulations and DFT calculations were instrumental in understanding the wear mechanisms, 42 which contributed to the observed tribological behavior. In future studies, the combination of MD simulations and DFT calculations should be focused on how the molecular–level insights from the simulations could complement the experimental data to more predict the COF and wear rate of hot–pressed nylon–66 samples under the different processing conditions.
Conclusions
(1) The hot–pressing time plays the significant role in enhancing the polymer chain rearrangement, lamellar alignment and microstructural densification, and the performance of hot–pressed nylon–66 samples is further improved, which is consistent with the results of MD simulations and DFT calculations. (2) The extended hot–pressing time enhances the amide–related bonding and hydrogen bonding of hot–pressed nylon–66 samples through the chain reorientation and improved molecular packing, as confirmed by the DFT and experimental results (3) The DFT calculations identify the key hydrogen–bond–active regions in the nylon–66, and the MD simulations further show that the hot–pressing time affects the chain mobility, packing density and stability, offering the valuable atomic–level insights into the improved properties of nylon–66. (4) The tensile strength and ductility of hot–pressed nylon–66 samples are increased with the hot–pressing time, and the uniform deformation enhances its tribological stability, which confirm the importance of hot–pressing time in enhancing the mechanical and tribological properties by the MD simulations (5) The MD simulations and DFT calculations provide the valuable atomic–level insights of hot–pressed nylon–66 samples, which provide the comprehensive understanding of its macroscopic mechanical properties and tribological behavior.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (24KJB430035), Nantong Social Livelihood Science and Technology Project (MSZ2024064, MS2025075).
Data Availability Statement
The datasets used and analyzed during the current study available from the corresponding author on reasonable request.