
Abstract
Lithium metal is a critical material in advancing energy storage technologies, particularly for next-generation lithium metal batteries (LMBs). However, industrial lithium metal production relies predominantly on LiCl–KCl electrolysis. This process faces several challenges and limitations, such as the need for high-purity LiCl, low current efficiency, and the generation of chlorine (Cl2) gas as a by-product. To ensure a sustainable and affordable lithium supply, recent studies have focused on improving process efficiency and developing alternative production routes. Despite this accelerating research activity, the lack of a unified and critical assessment of lithium metal production technologies hinders informed technology selection and scale-up. This review addresses this gap by examining recent advances in lithium metal production technologies, with a focus on improvements to conventional molten salt electrolysis, including feedstock optimisations, electrolyte modifications, and cell design innovations aimed at enhancing yield, purity, and energy efficiency. In addition, alternative low-temperature electrolysis enabled by aqueous-based lithium compounds, innovative electrochemical extraction methods, and thermochemical approaches using carbon and metallic reductants are summarised. The review also considers process–structure–performance relationships, highlighting how production conditions influence lithium metal microstructure and electrochemical properties. Finally, the review discusses the challenges associated with each method and highlights potential improvements to achieve more sustainable large-scale lithium metal production.
Introduction
Lithium, the lightest metal and the least dense solid element (0.53 g/cm3), possesses unique chemical and physical properties that have positioned it as a key material in modern technology. It is highly reactive, readily forming compounds with oxygen, nitrogen, and water, and exhibits excellent electrochemical activity due to its low standard electrode potential (−3.04 V vs SHE). 1 Lithium also demonstrates stable performance across a broad range of temperatures, thanks to its wide temperature window between its melting point (180.5 °C) and boiling point (1342 °C). 2 These characteristics make lithium critical for various applications. The addition of lithium to aluminium can reduce density while increasing the modulus of elasticity, which is highly beneficial for aerospace applications. 3 In pharmaceuticals, lithium is a primary standard in the treatment of bipolar disorder and has shown considerable antiviral potential, particularly in the management of COVID-19. 4 Moreover, due to its reactive nature, lithium has been used extensively in rocket fuel. The Li–SF6 combustion system can provide high specific energy (∼14.1 MJ/kg) and long-term energy storage, capable of powering space missions while providing both electricity and heat. 5
Since the early 2000s, the demand for lithium has increased significantly with the rise of rechargeable battery technologies. 6 Currently, lithium-ion batteries (LIBs) dominate the global rechargeable battery market, but several limitations make them less suitable for next-generation applications. 7 One of the main limitations is the utilisation of graphite anodes, which have relatively low energy density and theoretical capacity. 8 Moreover, the flammability of liquid electrolytes used in LIBs can lead to fire or explosion under overheating conditions, raising safety concerns for future usage. 9 To address these issues, lithium metal batteries (LMBs), which use lithium metal as the anode, are considered a promising replacement due to their significantly higher theoretical capacity and lower electrochemical potential. 10 However, the successful deployment of LMBs depends on the scalable production of high-purity lithium metal. As of 2024, global lithium metal production accounted for only 3% of the total lithium produced (240,000 metric tons). 11 It is predicted that demand will surpass the supply by 2026, signalling a critical bottleneck for future energy storage systems. 12
To meet this growing demand, more efficient production methods are required, yet the current industrial approach primarily relies on molten salt electrolysis of lithium chloride (LiCl). Although this method is well-established, it poses several significant drawbacks. One major limitation is the exclusive use of LiCl as the precursor, which restricts feedstock flexibility. Another is the moisture-sensitive nature of LiCl, which requires controlled conditions for storing and handling. The production of LiCl itself involves a tedious and energy-intensive process. First, ores containing spodumene (LiAl(SiO3)2) must first be converted into lithium carbonate (Li2CO3) by concentration using froth flotation, followed by high temperature calcination and acid or alkali digestion, and then a step of purification. 13 Subsequently, the obtained Li2CO3 is reacted with hydrochloric acid (HCl) to produce LiCl. 14 In addition, the electrolysis of LiCl generates hazardous chlorine gas (Cl2), which can reach up to 5 kg per kg of lithium metal produced, raising serious safety and environmental concerns. Furthermore, the method suffers from relatively low energy efficiency and high operational costs, posing challenges to its economic sustainability in meeting future lithium metal demand. 15 Figure 1 provides an overview of lithium metal production pathways from different feedstocks, highlighting industrial and alternative routes.
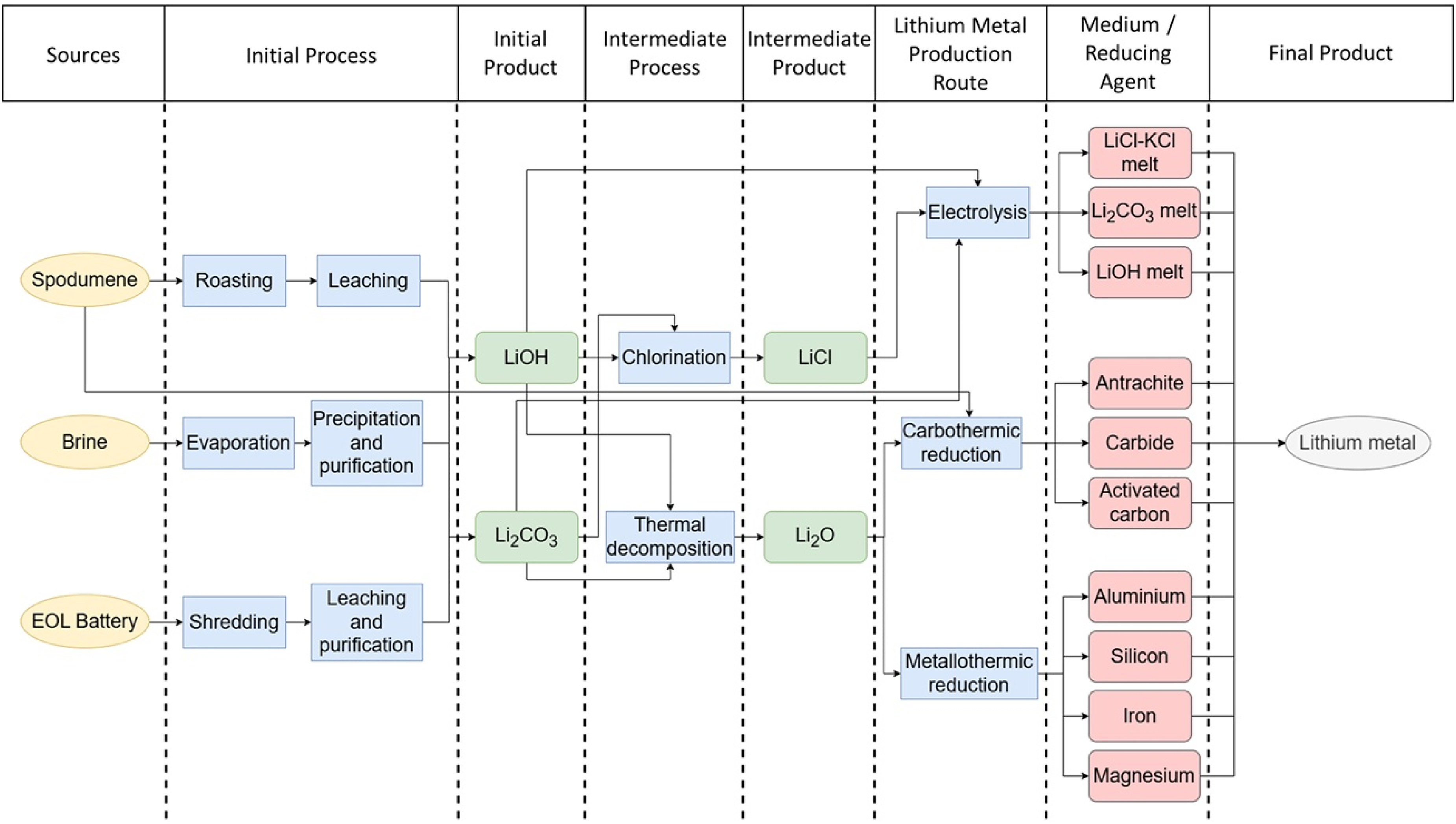
Integrated process map for lithium metal production from various lithium sources.
Thermochemical reduction methods, including carbothermic and metallothermic reduction of lithium-bearing compounds, have emerged as favourable alternatives to address these limitations. These approaches potentially allow the use of a broader range of lithium feedstocks and offer simplified processing routes that could reduce energy consumption and cost. Carbothermic reduction has been widely used for metal production16–18 and remains a benchmark process, whereas metallothermic reduction offers advantages such as lower operating temperatures due to its exothermic nature and the absence of environmentally harmful gaseous by-products. 19 However, both approaches face significant challenges because carbothermic reduction requires very high temperatures or vacuum conditions and produces CO or CO2 emissions. In contrast, the high cost and limited recyclability of metal reductants restrict further upscaling of metallothermic reduction. Hence, optimising reaction parameters, enhancing lithium recovery and purity, minimising environmental impacts from by-products, and scaling these processes are important for future development.
Recently, alternative methods such as the use of lithium-ion-conductive (LIC) ceramics have gained increasing attention. This approach is motivated by the abundant lithium content in brines, which, if directly converted into lithium metal, could significantly shorten the processing steps compared to those required for hard rock sources such as spodumene.20,21 The use of LIC as a membrane to separate the cathode and anode can effectively prevent the formation of impurities in the obtained lithium.22,23 However, further development remains challenging, especially in harsh brine conditions, where high salinity, temperature fluctuations, and competing ions can cause degradation or fouling. 24 Although direct lithium-metal extraction from brines is still at the laboratory scale, similar studies on extraction-to-salt processes suggest that the costs of membrane or separation systems could be prohibitively high. 25
This work is part of a broader research effort on the development of metallisation routes of lithium in the authors’ group and is presented in a series of papers. While previous review articles have primarily focused on lithium extraction from mineral and brine resources, molten salt electrolysis technologies, or the application of lithium metal in battery systems, comparatively limited attention has been given to a comprehensive analysis of lithium metal production routes themselves.
This paper, therefore, presents an in-depth review and discussion of both industrial and emerging methods for producing high-purity lithium metal. In contrast to the recent comprehensive review by Wang et al., 26 which focuses on lithium compounds and broader production and recycling systems, this work places greater emphasis on metallic lithium production routes and their underlying mechanisms. It examines the processes and equipment currently used in industrial electrolysis, together with strategies to optimise performance through improvements in cell design and raw material modifications. Furthermore, based on the gaps identified in previous work, 27 this review explores thermochemical routes and innovative electrochemical extraction in greater depth, including reductant selection, reaction mechanisms, process parameters, and product characterisation, while highlighting process–structure–performance relationships. Finally, the paper outlines current challenges and highlights opportunities for innovation, aiming to guide future research toward scalable, sustainable, and economically viable lithium metal production.
Industrial method for lithium metal production
Historically, lithium metal was first obtained in 1818 by William Thomas Brande and Sir Humphry Davy through the electrolysis of lithium oxide (Li2O). However, the amount produced was extremely small and highly reactive, igniting spontaneously upon exposure to air. 28 A significant breakthrough occurred in 1855, when August Matthiessen and Robert Bunsen successfully isolated lithium metal in larger quantities via the electrolysis of LiCl. 29 Despite this improvement, the electrolysis process at that time required very high operating temperatures due to the high melting point of LiCl (around 610 °C). To address this challenge, in the early twentieth century, Guntz proposed a eutectic mixture of lithium chloride and potassium chloride (KCl). 27 This eutectic system could lower the melting point of the electrolyte mixture to 380 °C, enabling the electrolysis to be carried out at more manageable temperatures (typically around 400–500 °C). This advancement allowed Metallgesellschaft AG to begin industrial-scale production in 1923, a method that remains the global standard for lithium metal production today.
The electrolysis process for producing lithium metal relies on the electrochemical decomposition of LiCl, in which lithium ions (Li+) migrate toward the cathode and are reduced to form liquid lithium metal. Meanwhile, chloride ions (Cl−) are oxidised at the anode to generate Cl2 gas. The reactions occurring in the cell can be represented by equations (1) to (3) as follows:
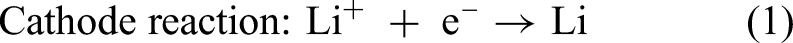

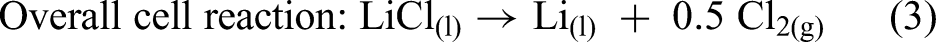
The eutectic mixture of LiCl-KCl plays a crucial role in reducing the melting point of the electrolyte, thereby improving ionic conductivity and reducing thermal degradation of cell components. 30 As shown in Figure 2, the eutectic composition occurs at approximately a 59:41 molar ratio of LiCl to KCl, which corresponds to the lowest melting point in the binary system. The lithium produced has a low density and floats on the surface of the molten salt and is then collected by mechanical skimming. The Cl2 gas evolved at the anode is toxic and corrosive; hence, it requires careful handling and removal.
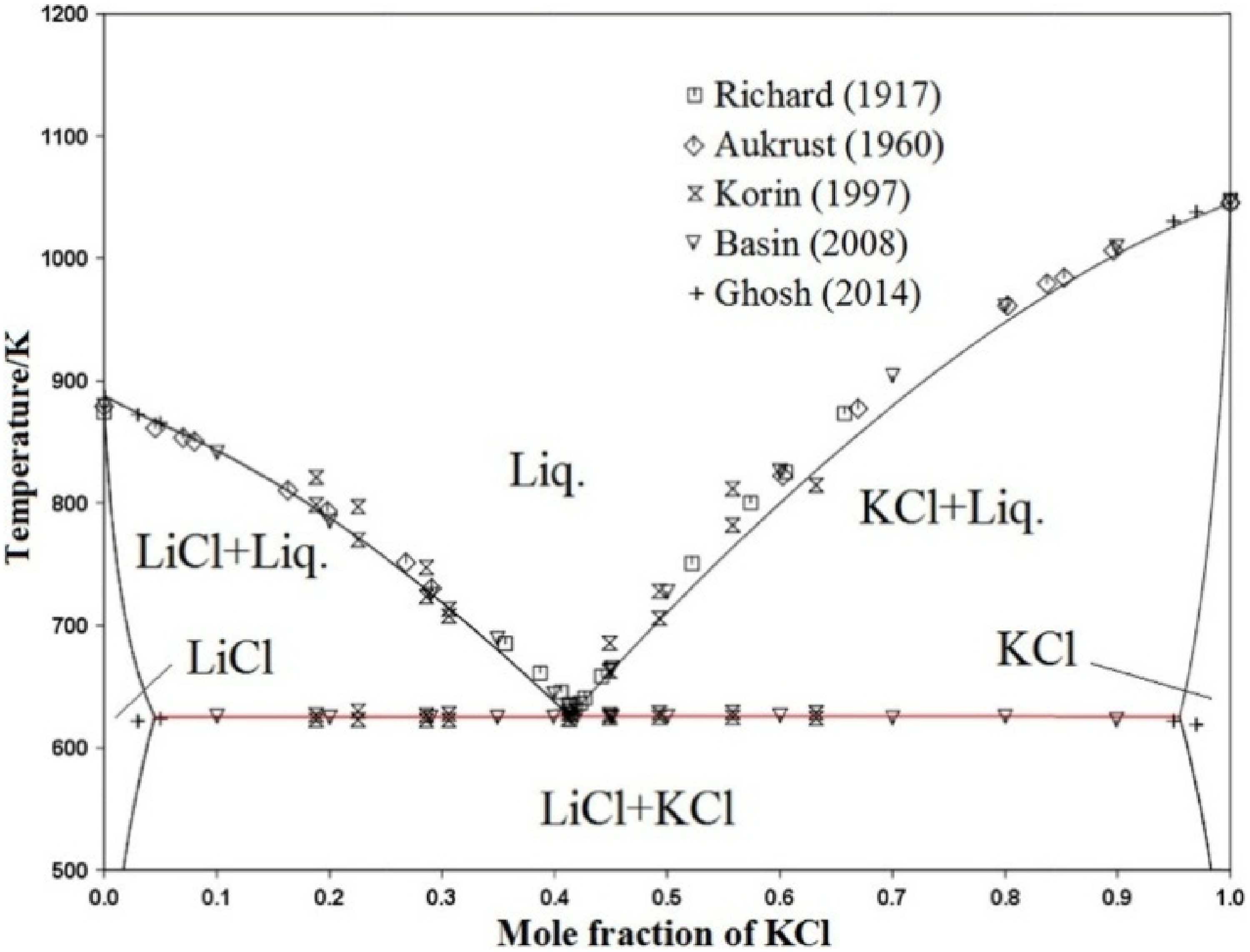
Phase diagram of LiCl–KCl mixtures. Reproduced from Ref. 31
Based on these fundamental electrochemical principles, industrial-scale lithium production employs specially designed electrolytic cells that optimise temperature control, material durability, and product collection to ensure efficient and safe operation. The electrolysis cell, as shown in Figure 3, typically consists of ceramic- or steel-lined crucibles that contain the molten LiCl-KCl electrolyte, maintained at 400–500 °C to keep the salts in a molten state. 32 An inert atmosphere, often argon gas, is maintained above the electrolyte surface to prevent lithium oxidation and contamination. 27 The cathode is usually made from steel or molybdenum, facilitating the reduction of lithium ions to liquid lithium metal. Meanwhile, the anode is commonly composed of graphite because of its good stability under Cl2 gas evolution. 33 To prevent the recombination of lithium and Cl2 gas, the cell design often incorporates iron gauze barriers. 34 The Cl2 gas generated at the anode is collected via gas outlet systems and subsequently processed or utilised in other industrial applications.
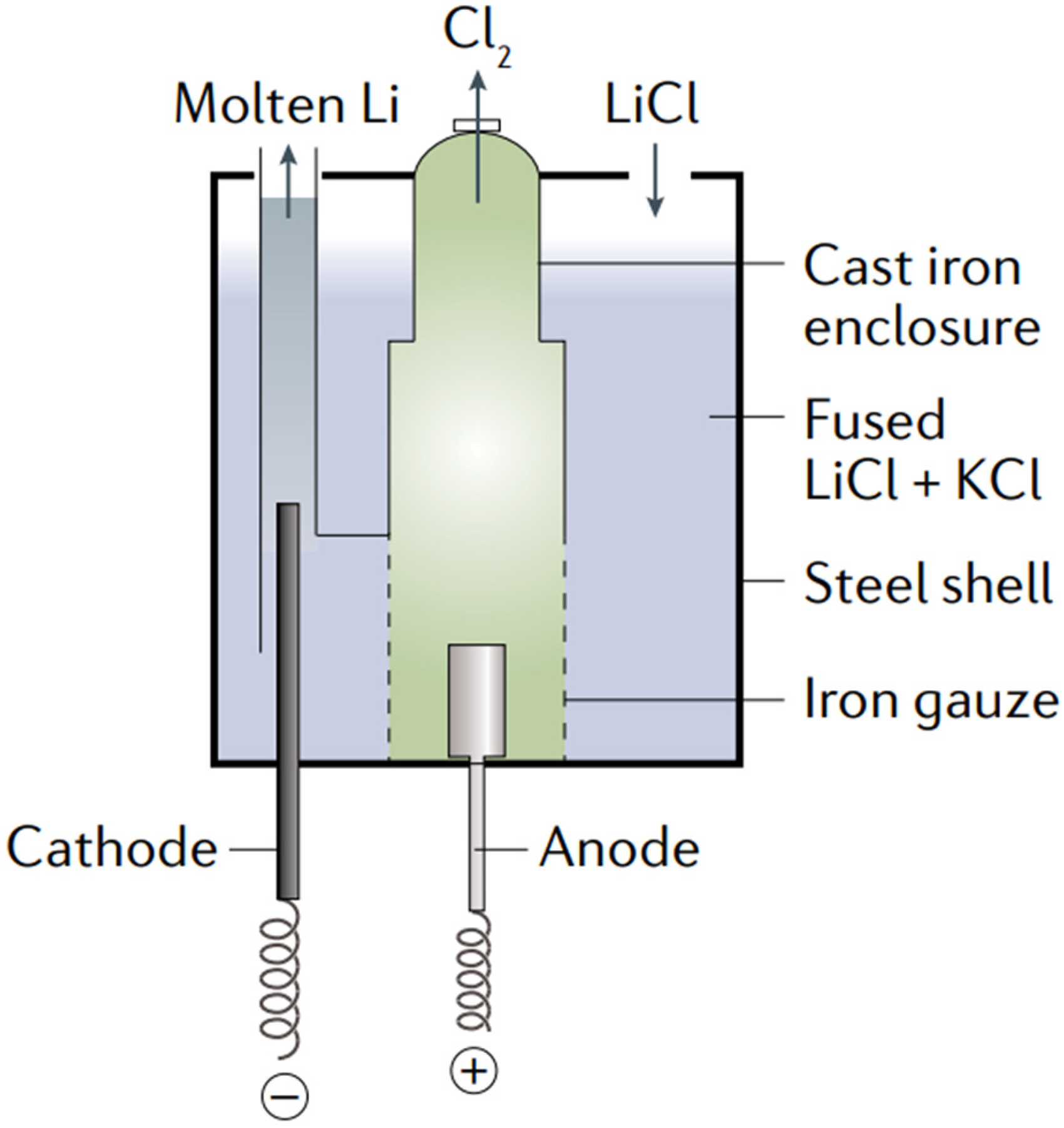
Typical electrolysis cell for lithium metal production. Reproduced from Ref. 34
Nevertheless, the electrolysis process for lithium metal production still faces several challenges, including the limited types of electrolytes that can be used, its relatively low energy efficiency of around 50%, and the generation of Cl2 gas as a by-product, which poses serious environmental hazards. To address these issues, many researchers have introduced innovations to modify several aspects of the electrolysis process, such as replacing the electrolyte and raw materials with safer alternatives that operate at lower temperatures, and redesigning the cell and its configuration to improve overall efficiency. These developments are discussed in the following section.
Development of the current industrial method
Electrolyte and raw material innovations
One major challenge in industrial lithium metal production via electrolysis is the exclusive use of LiCl as feedstock, which is highly hygroscopic and requires significant energy to produce. To tackle this issue, several studies have been conducted to reduce or replace LiCl by using alternative lithium compounds, including Li2CO3, Li2O, and lithium hydroxide (LiOH) in electrolysis cells as feedstocks or by using them directly as electrolytes.
Extensive research has focused on using Li2CO3 as a lithium source, which is not hygroscopic and consumes less energy overall because it avoids the conversion to LiCl. For instance, Foote Mineral Company patented a new method that utilises the Cl2 gas generated at the anodic compartment to react with Li compounds (such as LiOH and Li2CO3) at high temperatures to produce LiCl (equation (4)).
35
This approach can create a continuous process and reduce the utilisation of LiCl. However, the patent does not include a schematic showing how the system should be established.
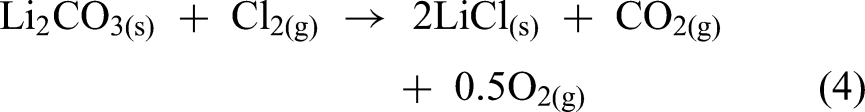
Similarly, DeYoung added Li2CO3 to the LiCl-KCl electrolyte before and during the electrolysis process. It was observed that after the addition, the cell voltage dropped, indicating that lithium carbonate was directly electrolysed as in equation (5). The absence of Cl2 gas formation further confirmed this. In contrast, when the Li2CO3 is directly mixed with LiCl to form an electrolyte, a black sludge composed of Li2CO3 and C was formed with only 5.8% lithium yield (equation (6)).
36
Kruesi and Fray observed a similar outcome when testing a pure Li2CO3–K2CO3 melt using a zinc solvent cathode to lower lithium activity. The lithium recovery was very low (∼6.4% efficiency) while carbon deposition dominated (∼89.5%), confirming that carbonate ion reduction strongly competes with lithium deposition in such systems.
37
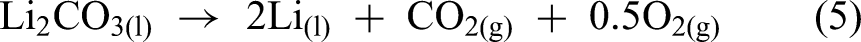
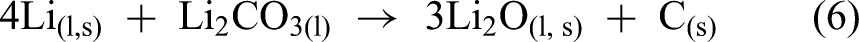
In a different approach, Sintim-Damoa et al. proposed an electrolytic process where Li2O, obtained by thermal decomposition of Li2CO3, is dispersed in a fluoride-based electrolyte (LiF–CaF2). 38 Then, the mixture was electro-reduced into lithium and absorbed in a liquid metal cathode. Li2O offers several advantages, including a lower decomposition potential (∼2.17 V) than LiCl, no Cl2 evolution at the anode, and it dissolves well in fluoride electrolytes. Besides, the LiF-CaF2 electrolyte also exhibits high chemical stability and low volatility, which serves as an efficient and safe electrolyte. However, the operating temperature for this system is much higher (750–900 °C) than the conventional electrolysis of LiCl-KCl, demanding more energy for its operation. Moreover, early experiments reported low efficiencies (∼20%), although improvements up to 80% efficiencies were anticipated with further optimisation.
More recently, LiOH has been investigated as a potential alternative to LiCl for lithium electrowinning, primarily to eliminate the costly conversion step from hydroxide to chloride. Takeda et al. attempted to use a LiOH–LiCl electrolyte with a ratio of 58:42 mol%.
39
After the electrolysis process was carried out, no lithium metal was recovered. Instead, a white deposit formed at the cathode, identified as a mixture of LiOH, LiCl, and Li2O. To further rationalise the failure of lithium deposition and the formation of these by-products from a thermodynamic perspective, the electrochemical stability relationships among the relevant species can be examined using the potential–pH diagram shown in Figure 4. Based on this diagram, the stability region of Li is located far from that of LiOH, suggesting that metallic Li is thermodynamically unstable in the presence of LiOH. Consequently, further reactions may occur, as described in equations (7) and (8).
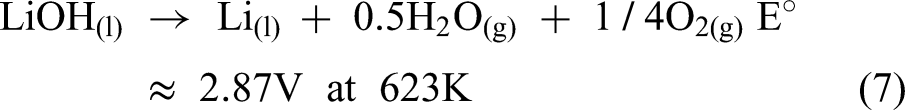
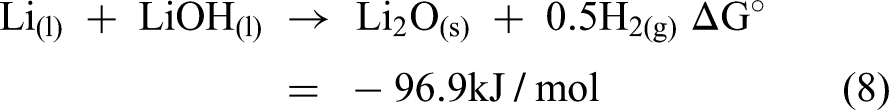
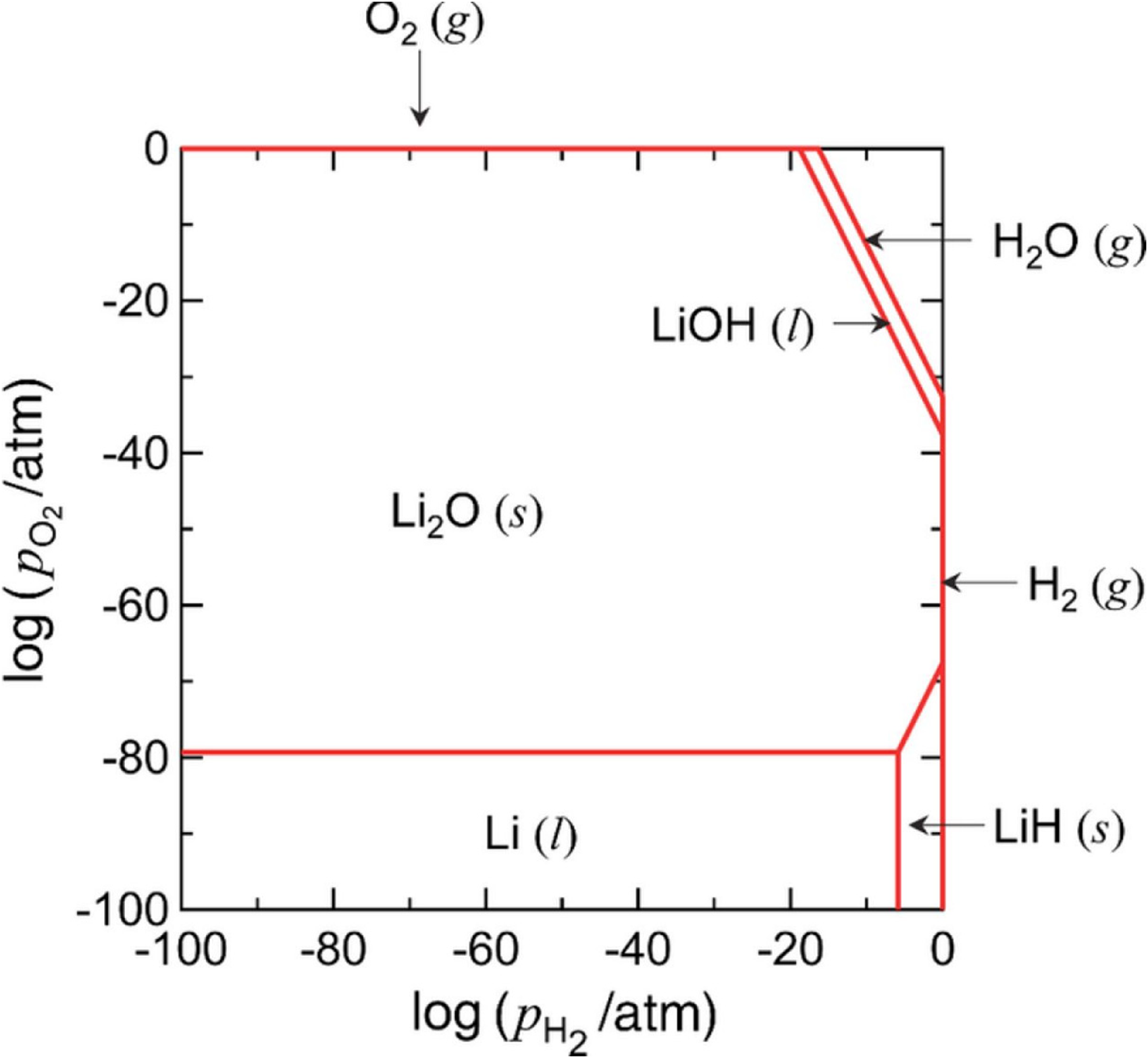
Potential diagram for the Li−H−O system at 673 K. Reproduced from Ref. 40
To overcome this problem, Takeda et al. incorporated LiOH as a feedstock in molten LiCl-KCl, where it was inserted into an anode compartment after the Cl2 gas was generated at the early stage of electrolysis. 39 This approach successfully prevents the back-reaction between lithium metal and LiOH and can produce lithium metal on the cathode, proving the feasibility of LiOH-based electrowinning. Likewise, Laude et al. utilised LiOH–LiCl electrolyte with a ratio of 70:30 mol% and could only obtain lithium metal at the cathode with a current efficiency of 37.9%. 41 These phenomena highlight the inherent challenges of using LiOH as an electrolyte: not only does lithium metal tend to react with LiOH to form Li2O and H2, but water migration from the anode to the cathode can also lead to re-oxidation of the deposited lithium, further reducing efficiency.
In an alternative way, Takeda et al. demonstrated that adding CsCl to LiCl–KCl eutectics significantly lowered the liquidus temperature, thanks to the large ionic radius of Cs, which disrupts the lattice structure. 40 This advantage enables the electrolysis at reduced temperatures (from 623 K to 548 K). Importantly, these improvements were achieved without sacrificing current efficiency, which remained high at 83.8–85.5%, comparable to LiCl–KCl systems. Furthermore, it can also suppress CO2 evolution at the graphite anode, which was proven by only 0.2% CO2 generation at 548 K.
Recently, a patent proposed the formation of anti-perovskite crystalline structure of lithium hydroxyhalide compounds (Li2OHX, where X = Cl, Br, or I), such as dilithium hydroxide chloride (Li2OHCl), by mixing LiOH with LiX (X = Cl, Br) to replace conventional electrolytes. 42 These compounds melt below 350 °C, nearly 100 °C lower than conventional LiCl–KCl systems. Besides that, Li2OHCl offers a higher lithium content per unit mass than LiCl, improving material efficiency. The process also runs at low voltage (<5 V) and generates oxygen and hydrogen halides (HX, where X = Cl, Br, I) instead of Cl2 gas. When the electrolysis was conducted for 50 h, the metallic lithium droplets were formed with a purity exceeding 99.5%.
Cell design and configuration innovations
The configuration of the electrolytic cell is crucial in determining process efficiency, product purity, and operational stability. Currently, the challenges in industrial cell designs include high energy consumption, poor separation of anodic and cathodic products, and limited control over lithium deposition behaviour. To address these issues, recent innovations have focused on redesigning the cell architecture and configuration, as summarised in Table 1. One of the most notable innovations in the design of electrolytic cells for lithium metal production is the replacement of solid cathodes with liquid metal cathodes, particularly liquid tin. Tin serves as an efficient liquid cathode because it can form an alloy with lithium. This alloy formation prevents lithium from floating on the electrolyte due to the high density of tin (7.3 g/cm3), thus preventing reactions with other gases such as oxygen or chlorine. Furthermore, tin is thermodynamically favourable with lithium (to form a Li-Sn intermetallic), allowing lithium to deposit at a more positive potential. 43 This phenomenon is advantageous because it reduces overall energy consumption. As first proposed by Sintim-Damoa et al., the concept involves the electroreduction of lithium compounds in a fused salt electrolyte, with lithium being absorbed into a liquid metal cathode. The liquid cathode not only facilitates efficient lithium separation from the electrolyte but also simplifies downstream purification through distillation or further electrolysis. 38
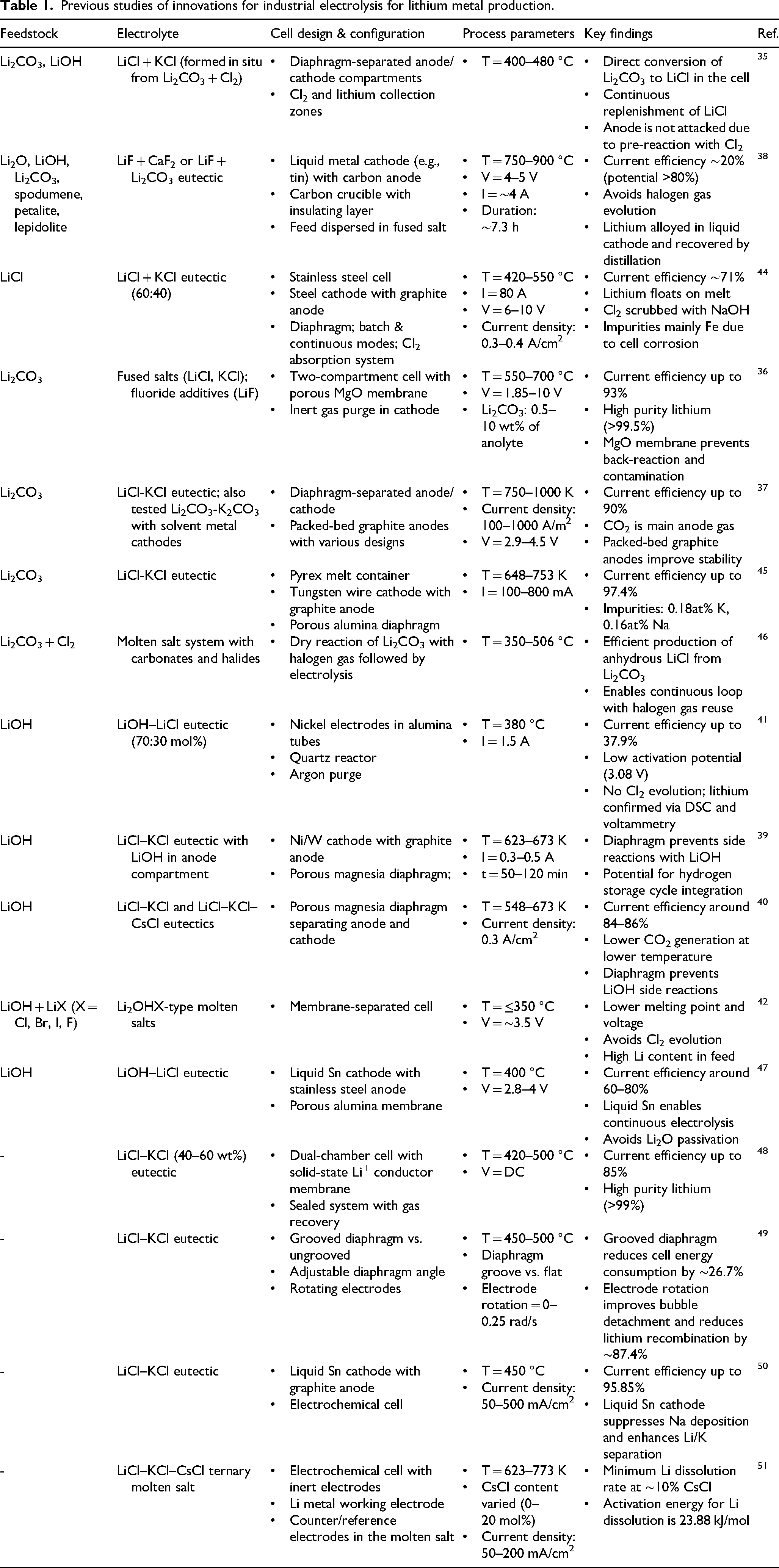
Previous studies of innovations for industrial electrolysis for lithium metal production.
Decades later, Tang and Guan demonstrated a modern implementation of this concept by employing a liquid tin cathode in molten LiOH–LiCl electrolysis, as depicted in Figure 5. 47 It was found that lithium could be effectively alloyed with tin, facilitating easier collection and reducing the risk of lithium passivation. Building on these innovations, Lei et al. further optimised the system by quantifying the electrodeposition efficiency and selectivity of lithium over common impurities (e.g., Na+, K+, Mg2+, Ca2+) in LiCl–KCl molten salts. 50 The results revealed a current efficiency of up to 95.85% at 500 mA/cm2, which is significantly higher than that of inert cathodes. The alloying effect of liquid metal can eliminate the formation of metal fog because the diffusion of Li2+ ions to the anode can be hindered, as shown in Figure 6.
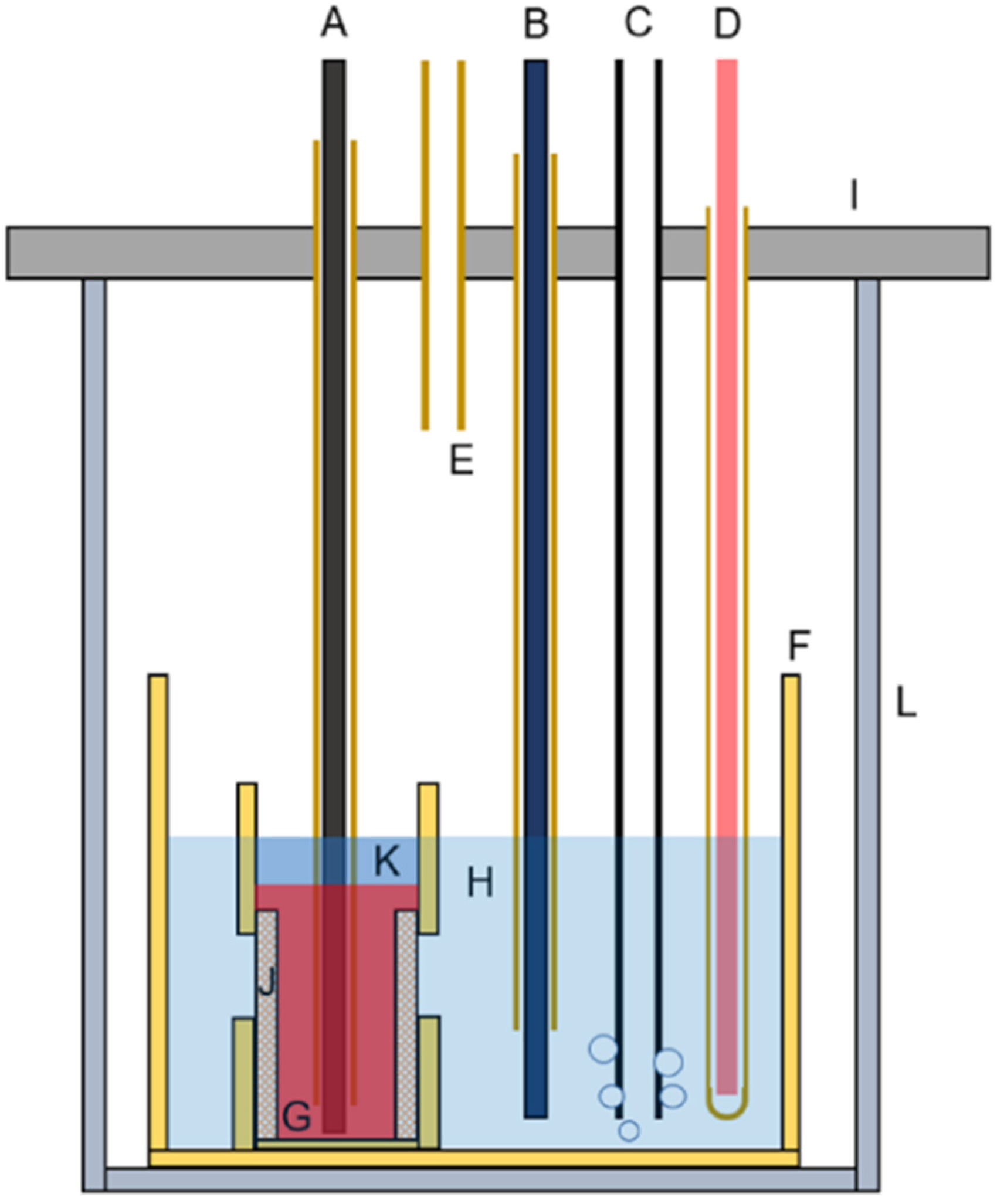
Schematic diagrams of the LiOH electrolysis cells A – tungsten current collector inside an alumina tube, B – nickel pseudo-reference electrode housed in an alumina tube, C – stainless-steel anode combined with a gas bubbling tube, D – thermocouple probe protected by a closed-end alumina tube, E – gas outlet, F – alumina crucible container, G – liquid tin cathode, H – molten LiOH–LiCl electrolyte, I – stainless-steel flange, J – porous alumina membrane joining the alumina crucible at the bottom and alumina tube at the top, K – molten eutectic LiCl–KCl, L – quartz tube. Reproduced from Ref. 47
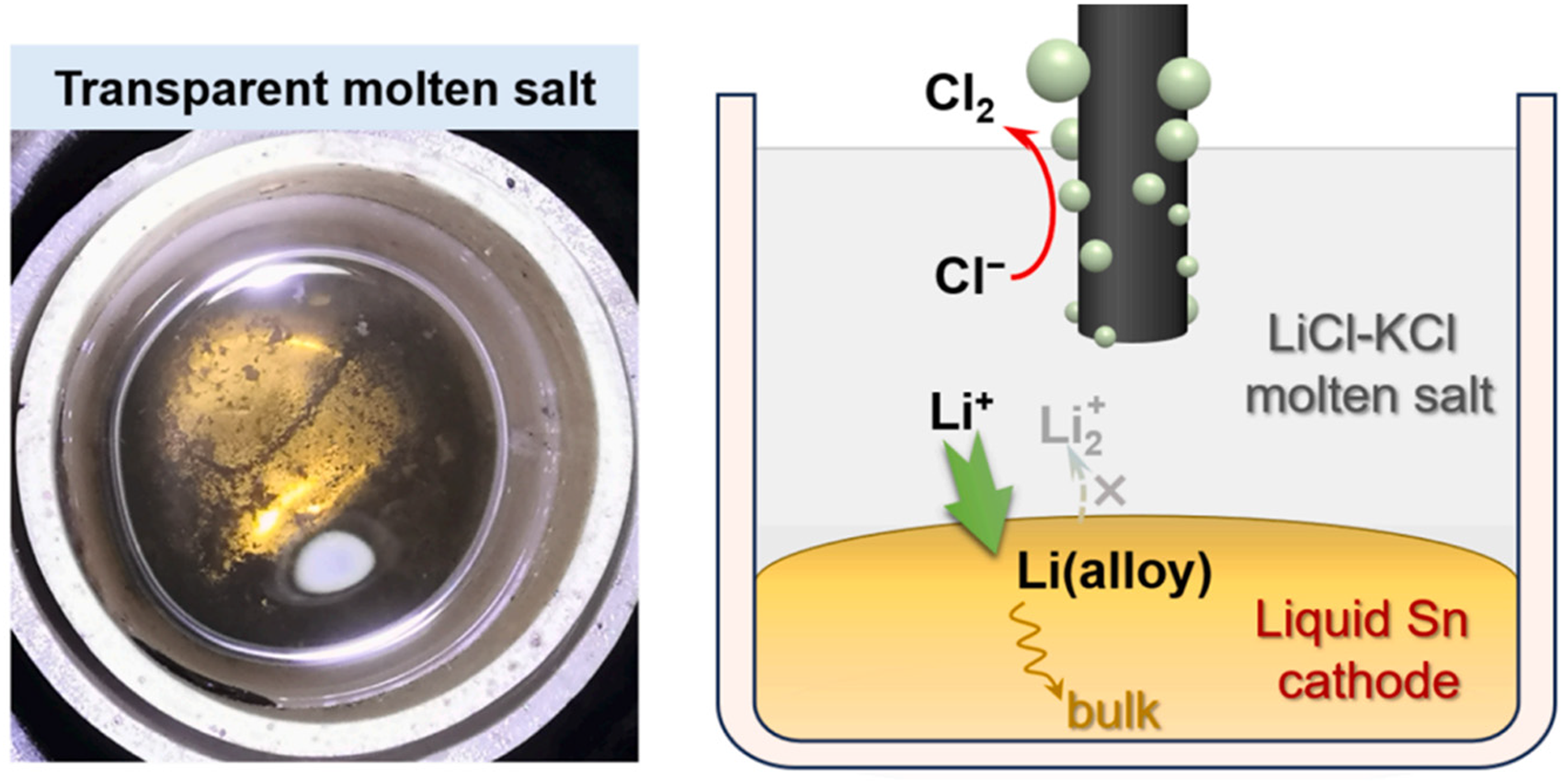
Digital photo and schematic diagram of liquid Sn cathode electrolysis. Reproduced from Ref. 50
Another critical innovation in lithium electrolysis cell design is the incorporation of diaphragms or membranes to separate the anode and cathode compartments. This configuration addresses a fundamental challenge in molten salt electrolysis: the undesired recombination of lithium metal with anodically evolved Cl2 gas, which can significantly reduce current efficiency and product purity. Early work by Hoh et al. demonstrated the use of a stainless steel diaphragm between the anode and cathode to reduce direct contact between the two products, but the system remained a single-compartment cell. 44 While this configuration improved lithium collection and achieved a current efficiency of up to 71%, it did not fully prevent the diffusion of Cl2 toward the cathode or the reoxidation of lithium, especially under continuous operation. To solve this issue, DeYoung's patent introduced a more advanced approach using porous magnesium oxide to separate the lithium carbonate-fed anode and cathode compartments. 36 This design not only prevented back-diffusion of lithium but also enabled direct electrolysis of lithium carbonate. Moreover, this design also allowed for independent control of gas environments, where argon was supplied over the cathode and air or inert gas over the anode, achieving current efficiencies of up to 93% and lithium purities exceeding 99.5%. Building on this, Takeda et al. implemented a two-compartment cell design using a porous magnesia diaphragm (20% porosity, 5 mm wall thickness) to prevent the back-reaction between lithium and LiOH (Figure 7). 40 Electrolysis conducted in both LiCl–KCl and LiCl–KCl–CsCl eutectic melts achieved cathode current efficiencies of 84–86%, suggesting the ability of the diaphragm to allow lithium ions to migrate from the anode side to the cathode while effectively restricting the transport of hydroxide ions.
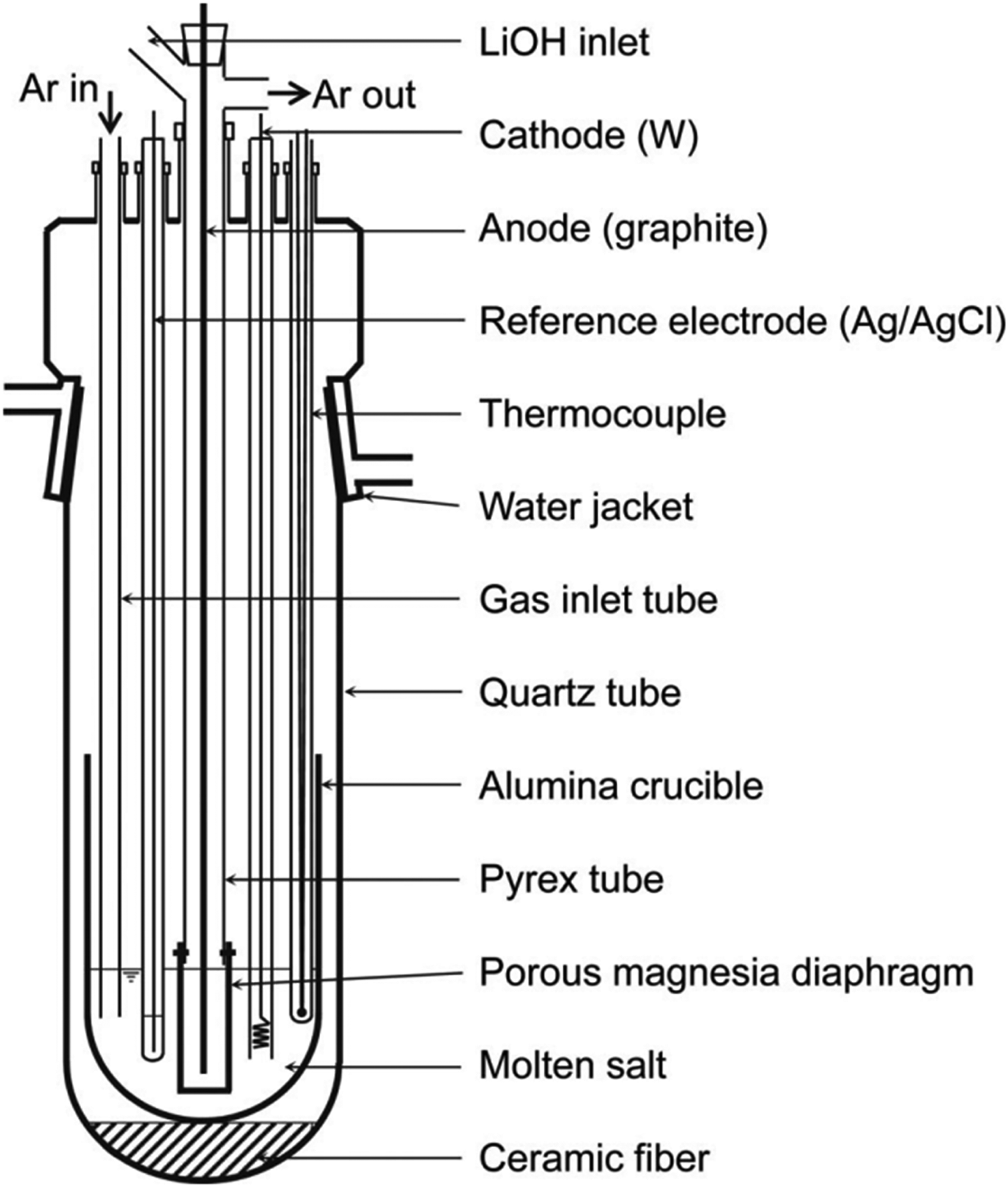
Experimental apparatus used to investigate the electrochemical behaviour of molten LiOH−LiCl and its electrolysis. Reproduced from Ref. 40
In a comprehensive study, Kruesi and Fray also evaluated the effects of the pore size of the diaphragm. 37 It was found that the refrasil (woven silica fibre cloth) diaphragm, due to its relatively large pores, allowed significant transport of carbonate ions from the anolyte to the catholyte, resulting in excessive carbon deposition and a low current efficiency of only 19%. In contrast, the porous alumina diaphragms demonstrated much better selectivity. Diffusion experiments confirmed that the carbonate ion diffusion coefficient was significantly lower for the 0.01 mm diaphragm (3.2 × 10−6 cm2/s) compared to the 0.1 mm diaphragm (9.2 × 10−6 cm2/s). However, this reduced pore size also affects the current efficiency achieved. The 0.1 mm diaphragm can achieve 48% current efficiency, while the smaller size (0.01 mm) can only reach up to 16%. This suggests that overly small pores may hinder lithium-ion transport, increasing cell resistance or limiting current flow. 37
In more recent developments, membrane-based separation has emerged as a promising alternative to traditional diaphragms for lithium electrowinning. A 2020 patent by Beijing University of Chemical Technology proposed the use of a membrane with micro- to nano-scale porosity (1 nm to 1 cm), made from materials such as porous ceramics, glass, stainless steel, or lithium-ion-conducting ceramics (e.g., LISICON, perovskite, or garnet-type structures). 42 This membrane can prevent the reoxidation of the lithium metal deposited in the cathode while also improving product purity, which can reach greater than 99.5%. Similarly, a patent from Central South University introduced a solid-state electrolyte membrane in a dual-chamber molten salt electrolysis cell for high-purity lithium production. 48 The system uses a solid lithium-ion-conducting ceramic membrane to separate the anode and cathode chambers. The membrane selectivity ensures that only lithium ions are transported, significantly enhancing product purity (up to 99%) and enabling a fully enclosed, automated process with improved energy efficiency and environmental safety.
Overall, the investigation of alternative lithium feedstocks reveals a persistent trade-off between material advantages and electrochemical limitations. Although Li2CO3 and LiOH reduce reliance on hygroscopic LiCl and simplify upstream processing, their direct use is hindered by competing reactions and thermodynamic instability that suppress lithium metal yield. Carbonate systems, for instance, favour carbon deposition over lithium recovery, while hydroxide-based electrolytes promote back-reactions that convert freshly deposited lithium into Li2O and H2. Even strategies such as in situ chlorination or controlled LiOH feeding only partially resolve these issues and often reintroduce dependence on chloride chemistry. Meanwhile, Li2O-based systems offer a promising chlorine-free pathway with favourable decomposition potentials, but their requirement for high operating temperatures and still-developing efficiencies limits immediate industrial applicability.
At the same time, innovations in electrolyte formulation and cell design have shown stronger potential to address these challenges from a systems perspective. Additives such as CsCl and emerging lithium hydroxyhalide electrolytes can lower operating temperatures and improve energy efficiency, though questions remain regarding long-term stability and scalability. More importantly, advances in cell architecture—particularly the use of liquid metal cathodes and ion-selective diaphragms or membranes—have significantly improved lithium recovery, purity, and process control by mitigating recombination and stabilising deposited lithium. However, these approaches introduce new engineering constraints, including mass transport limitations and material durability. Taken together, these findings suggest that no single innovation is sufficient; instead, future progress in lithium electrolysis will depend on integrating optimised feedstocks, tailored electrolytes, and advanced cell configurations to achieve a balance between efficiency, stability, and industrial feasibility.
Alternative method for lithium metal production
Aqueous low-temperature electrolysis
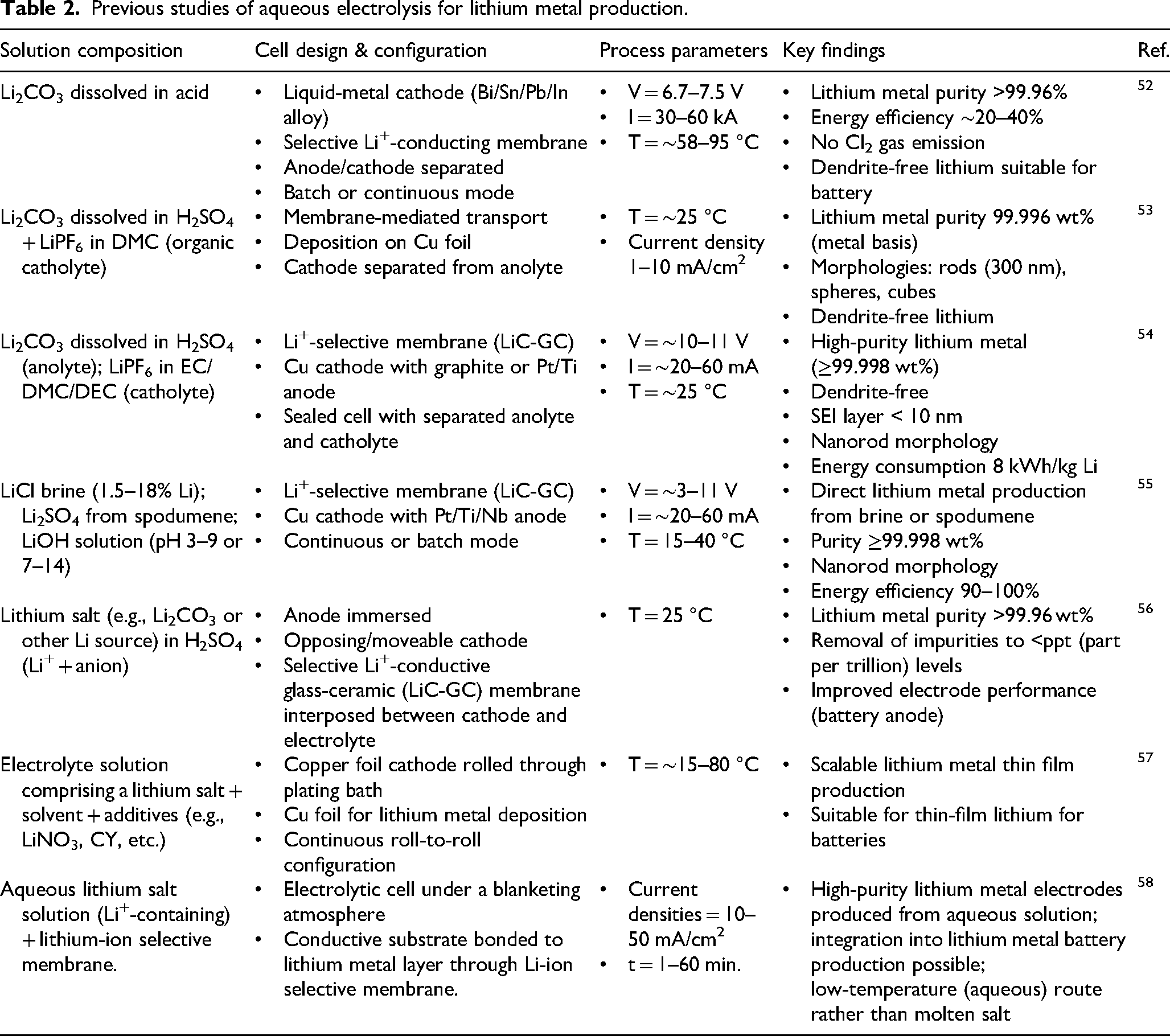
The aqueous low-temperature electrolysis approach has emerged as a promising alternative for replacing traditional high-temperature molten salt electrolysis. It is considered sustainable in terms of energy efficiency and environmental impact because it utilises water-based electrolytes, which can significantly reduce operational temperatures. Additionally, it can simplify system design and improve safety by avoiding extreme temperatures and corrosive molten salts. Table 2 compiles the studies that utilised low-temperature electrolysis of aqueous lithium sources for obtaining lithium metal.
Previous studies of aqueous electrolysis for lithium metal production.
Amendola proposed aqueous electrolysis below 100 °C to produce lithium metal. This is achieved by dissolving Li2CO3 salt or other lithium sources in water, aided by acids such as sulphuric acid or trifluoromethanesulfonic acid to enhance solubility and ion availability. 52 Electrolysis is performed at 1.7–1.95 V and currents between 0.1–0.45 A in a cell containing a liquid metal cathode, typically an alloy of bismuth, lead, tin, indium, or gallium. Lithium ions are reduced at the cathode to form a lithium-metal alloy. Further purification using distillation or chemical methods is then conducted to obtain pure lithium metal.
In a separate study, Mashtalir et al. demonstrated room-temperature electrolysis of an aqueous mineral solution using a two-compartment cell separated by a lithium-ion conducting glass ceramic (LIC-GC) membrane, as shown in Figure 8. 53 The anode side contained an aqueous solution, while the cathode side used an organic electrolyte (LiPF6 in DMC). This setup enabled morphological control (Figure 9) by adjusting current density between 1–10 mA/cm2, and produced lithium metal with 99.996 wt% purity and high crystallinity (Figure 10). Building on this, a scalable system was later developed, reaching up to 99.998 wt% purity and reducing energy consumption to just 8 kWh/kg, which is far lower than that of conventional molten salt electrolysis (30–35 kWh/kg). 54 Later, they advanced the system by employing a multi-layer composite barrier—typically combining a LiC-GC with an additional lithium-ion conductive barrier film (Li-BF). 56 In addition, the cathode design also allows for controlled deposition morphology, including nanorod structures. Through this combination, they can obtain lithium metal with purity up to 99.9997 wt% on a metal basis.
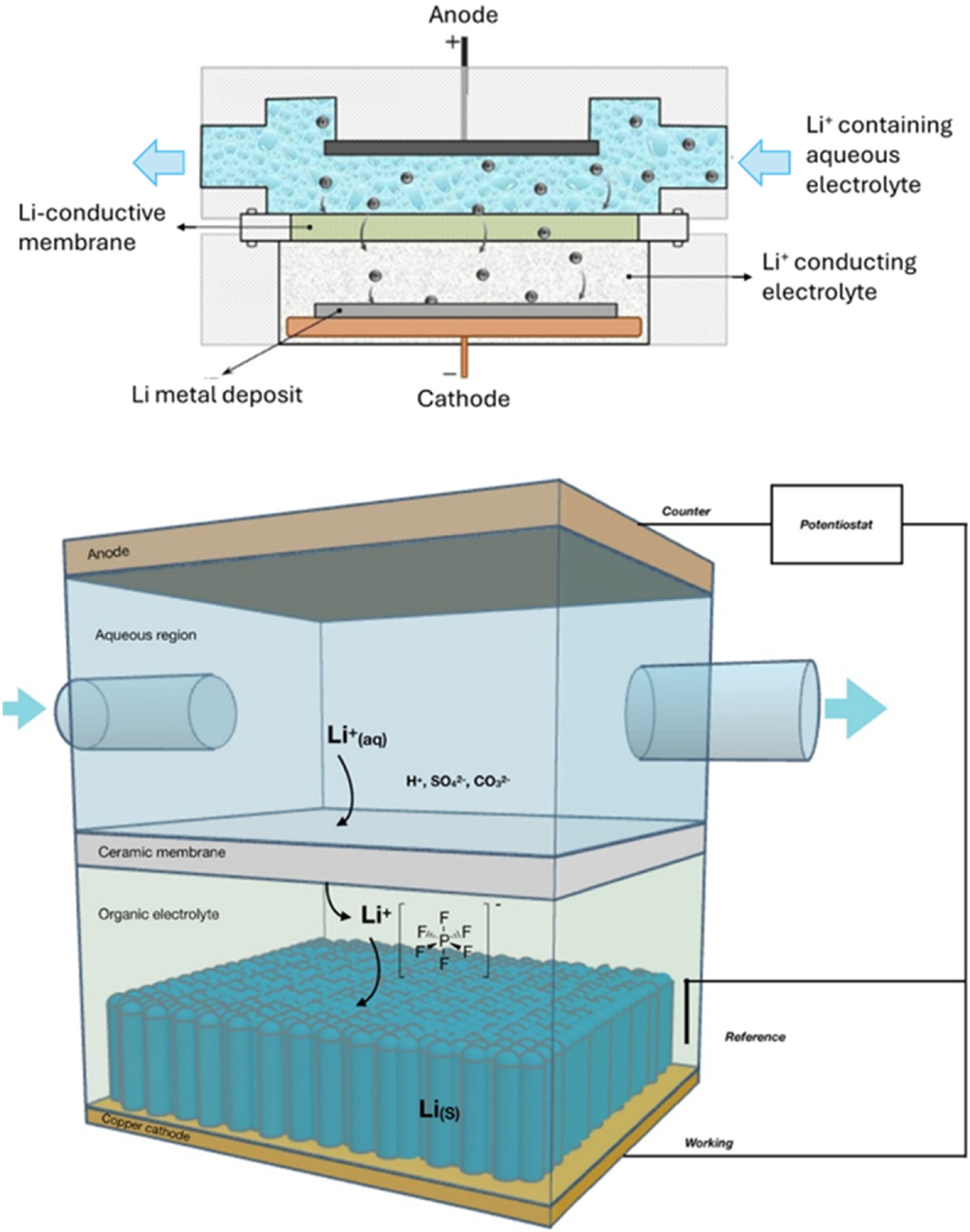
Schematic representations in two-dimensional and three-dimensional of the electrochemical cell. Adapted and reproduced from Ref. 53

SEM images of electrolytically deposited Li films with different morphology. Reproduced from Ref. 53
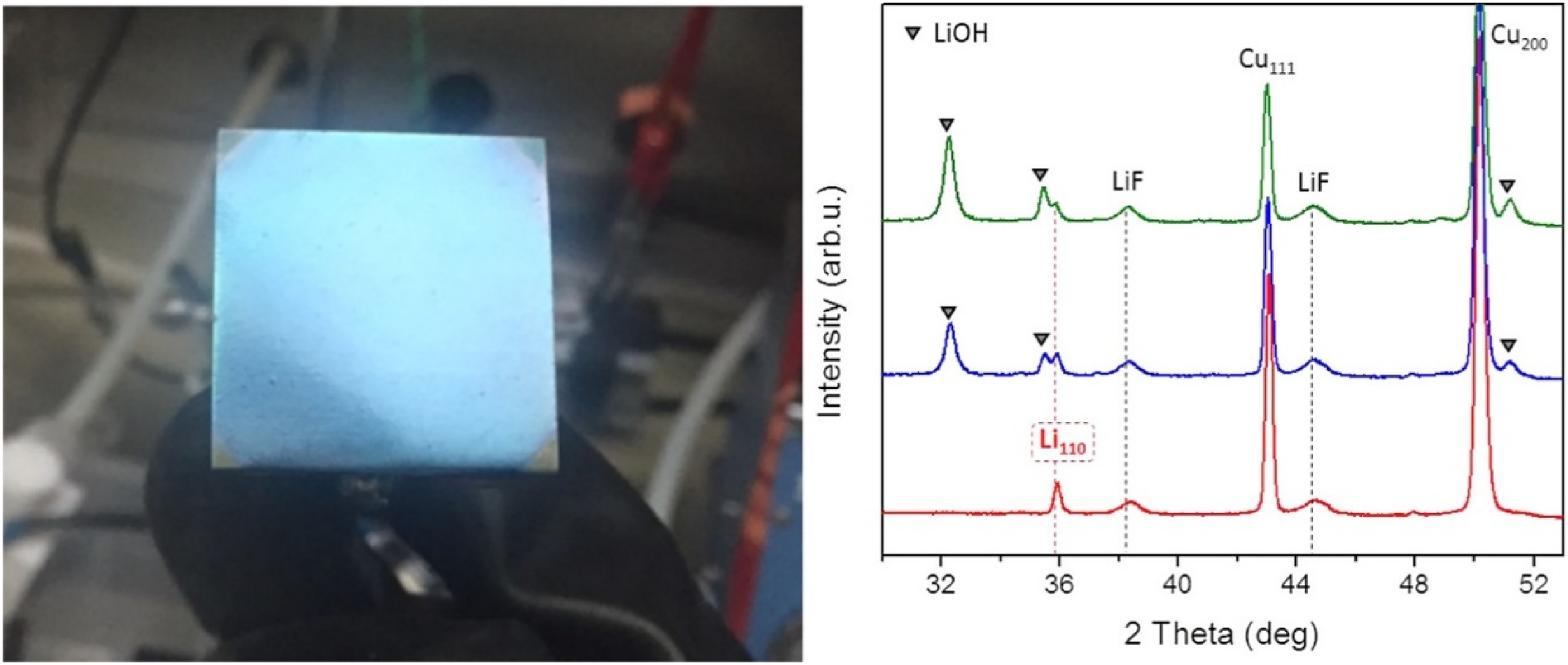
Pristine nanorod film under an argon atmosphere and X-ray diffractograms of the lithium film before (red) and after 15 and 30 min (blue and green, respectively) exposure to air. Reproduced from Ref. 53
In contrast, Alpha-En Corporation demonstrated direct lithium production from various aqueous sources, including lithium chloride brines (1.5–18% Li), LiOH solutions, and spodumene sulphate liquors, without intermediate conversion to lithium carbonate. 55 The cell design also utilised a lithium-ion-selective membrane to separate the anode and cathode. The process operates at low temperatures, typically between 15 °C and 40 °C, with an optimal temperature around 23 °C, while the voltage used is −3.75 V and the current is around 70 mA. In one example, using lithium chloride brine as the electrolyte and setting the electrolysis duration to 7200 s, they produced a blue-coloured lithium film, indicating a nanorod morphology. Meanwhile, decreasing the duration to 2500 s yielded a grey film with dense spherical morphology.
Recently, Donghyeon et al. developed a scalable method for lithium metal production via electrodeposition at room temperature (15–80 °C), utilising a two-compartment electrolytic cell separated by a lithium-ion conducting membrane. 57 This system enables lithium ions to migrate from an aqueous anolyte, typically composed of inexpensive lithium salts such as Li2CO3 or LiCl, into an organic catholyte where lithium metal is deposited onto a copper substrate.
Overall, aqueous electrochemical routes can operate at temperatures below 100 °C while producing lithium metal as thin films with tuneable morphologies and nanoscale particle sizes. Furthermore, the produced lithium can achieve very high purity (>99.99%), which is advantageous for lithium metal battery anodes, as electrowinning and anode manufacturing may potentially be integrated into a single process, thereby reducing downstream processing requirements. However, the practical significance of morphology control and direct film deposition remains uncertain at larger scales, where maintaining uniformity and performance becomes increasingly challenging. In addition, although the use of Li2CO3 as a feedstock provides economic advantages, its limited solubility necessitates acid-assisted dissolution (e.g., sulphuric acid), introducing additional reagent consumption, corrosion challenges, and waste generation, which may offset its cost benefits. Another drawback of low-temperature aqueous electrolysis is its relatively slow reaction kinetics compared with high temperature molten salt electrolysis, which must be considered for its suitability for larger scale industrial processes.
Therefore, despite the attractive low-temperature operation and high product quality, the overall industrial viability of aqueous electrochemical lithium production remains uncertain and will depend on successful process integration, efficient electrolyte management, and its ability to compete with established high-temperature electrochemical methods.
Innovative electrochemical extraction
Beyond low-temperature electrolysis, various innovative electrochemical approaches have also been developed, as summarised in Table 3. Early efforts to improve energy efficiency in lithium metal production involved hybrid electrochemical designs. For instance, Cooper et al. introduced the bipolar cell comprising an aqueous cell and a molten salt cell. In the aqueous cell, operated at 30–40 °C, lithium hydroxide (LiOH) is electrochemically reduced at the amalgam cathode to form a lithium-mercury amalgam. 59 The amalgam then flows into the molten salt cell, where lithium is stripped from the amalgam and deposited as pure metal in a LiI-CsI eutectic melt maintained at 225 °C. Although this configuration allows for high energy efficiency (>70%), the use of mercury remains a significant concern due to its toxicity and environmental risks.
Previous studies of innovation on electrochemical extraction for lithium metal production.
Separately, Lang et al. developed a novel system using low-grade molten LiCl (95 wt%) mixed with AlCl3 as the electrolyte. 15 The addition of AlCl3 was intended to lower the melting point of the mixture to 240 °C. Unlike conventional systems, AlCl3 cannot typically be used as an electrolyte component because aluminium would become an impurity in the produced lithium metal. To overcome this issue, a solid electrolyte made of Li6.5La3Zr1.5Ta0.5O12 (LLZTO) was employed, allowing only lithium ions to pass through. Furthermore, the system prevents Cl2 gas generation since an aluminium anode was used. This configuration achieved a lithium recovery of 82%, a coulombic efficiency of 96%, and a purity of 99.7%. Using this method, the cost of lithium metal production is estimated to be reduced by up to 20% based on 2020 market conditions. 15
A more advanced approach by Li-Metal Corp. introduced a flow-through electrowinning cell designed for lithium metal production from lithium carbonate or lithium hydroxide feedstocks. 64 This system utilised a porous ceramic membrane to separate the anolyte and catholyte chambers to minimise backflow of impurities. The cell operates at 400–600 °C and uses a hydrostatic pressure differential to control ion flux direction, which helps prevent membrane fouling and enhances current efficiency. A key innovation is the use of freeze-seal assemblies that form self-healing layers of frozen electrolyte, providing both fluidic and electrical isolation. In addition, the catholyte can include carrier metals (e.g., bismuth, tin, and indium) to form lithium alloys in situ, improving product stability and handling.
Besides electrochemical designs, recent focus has shifted to solid-state electrolytes and membrane-assisted electrochemical systems that eliminate liquid-phase instabilities. In 2018, Yang et al. introduced a lithium extraction technique using a solar-powered electrolysis device. 60 They designed a dual-compartment cell in which seawater served as the anolyte, and a non-aqueous catholyte was placed behind a lithium-ion-selective solid electrolyte membrane (a NASICON-type or Lithium Aluminium Germanium Phosphate/LAGP ceramic), as shown in Figure 11. The system reportedly achieved a lithium production rate of about 5.7 mg/dm2h at a current density of ∼240 µA/cm2, before side reactions (e.g., electrolyte decomposition) became dominant at higher current densities.
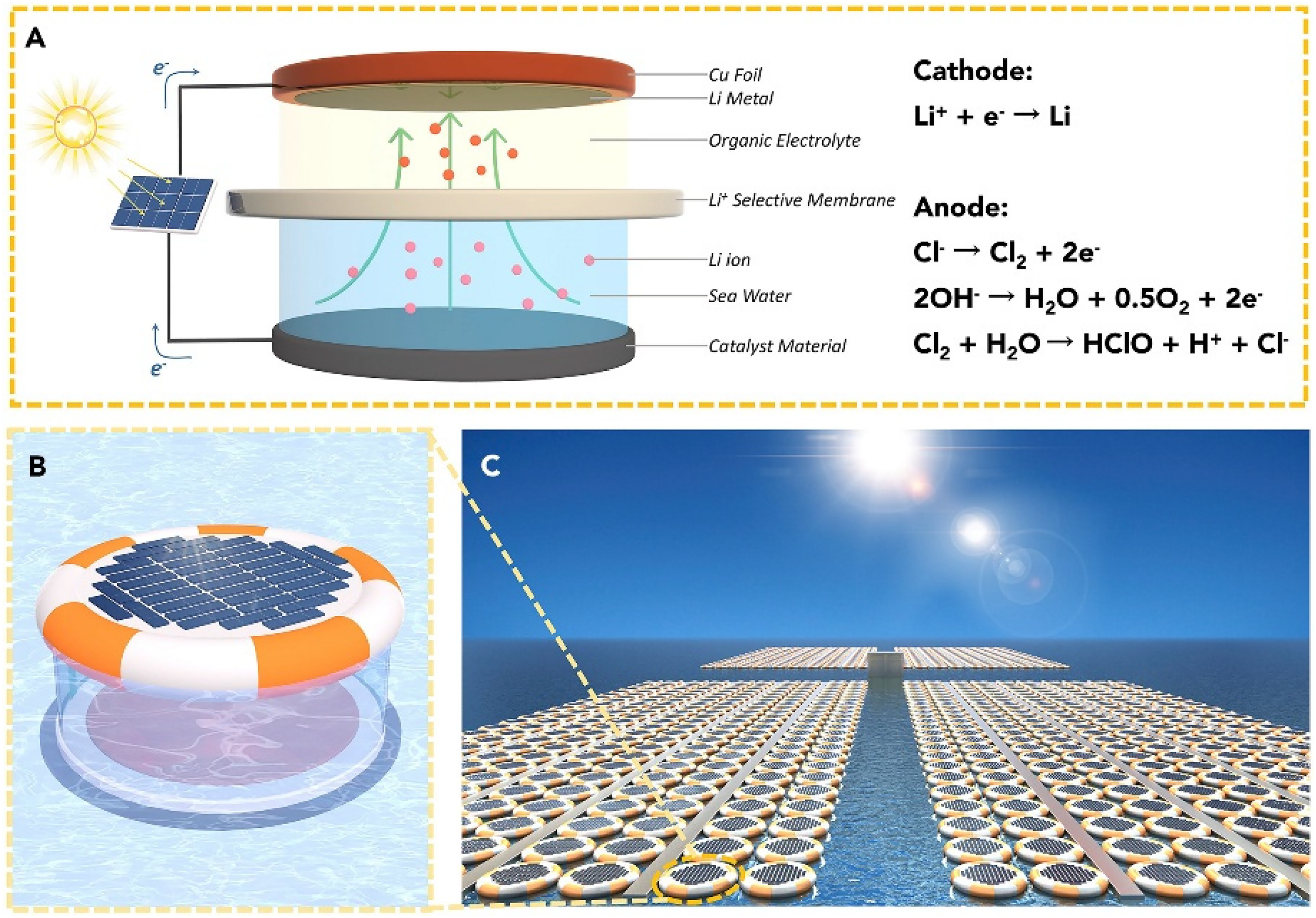
Schematic diagram of the solar-powered lithium extraction device. Reproduced from Ref. 60
Zhao et al. later developed a system with superior performance compared to conventional liquid electrolyte systems. 61 The device utilised a high-performance garnet-type solid-state electrolyte, Li7La3Zr2O12 (LLZO), synthesised via spark plasma sintering. The resulting device achieved an impressive lithium extraction efficiency of 198 μg/cm2/h in natural seawater. Economically, this process only required energy costs as low as $1.5–2.7/kg-Li in 2020, 61 based on reported energy consumption and industrial electricity prices in China (0.08–0.15 $/kWh), suggesting its potential as a lithium metal production technology for the future. Subsequently, Zhang et al. improved this system by applying a TiO2-modified garnet-type solid-state electrolyte (LLAZO) to overcome the critical challenge of poor interfacial contact between lithium metal and solid electrolytes. 62 The incorporation of a lithiated TiO2 interlayer significantly reduces interfacial resistance by up to three orders of magnitude, as confirmed by the absence of void space (Figure 12). Notably, although the reported extraction efficiency was only 110 μg/cm2/h, the device exhibited superior long-term stability, maintaining consistent performance over 40 cycles with minimal voltage fluctuation (±0.05 V).

Cross-sectional morphology of the garnet solid-state electrolyte/Li metal interface. Reproduced from Ref. 62
Further progress was achieved through direct electrolytic extraction of lithium (DEEL) using a sandwich-structured garnet-type solid electrolyte, LLZTO (Figure 13). 65 This type of solid electrolyte exhibits excellent water stability, which was previously a major limitation of conventional LLZO-based electrolytes. Furthermore, the LLZTO membrane features a lithium-deficient La2Zr2O7 surface layer that enhances moisture resistance while maintaining high ionic conductivity (7.5 × 10−4 S/cm) and low interfacial resistance (40 Ω·cm2). The DEEL system enables direct lithium metal extraction from both simulated and natural brines, including environments with high Mg/Li ratios (up to 130). Under these conditions, the system achieves a current efficiency of up to 97.71% and produces lithium metal with a purity of 99.48 wt%. Similarly, a recent patent from Jiangsu Xinliyuan Technology Co., Ltd describes a solid-state electrolysis system using garnet-type solid electrolytes such as LLZTO from low-purity lithium salts, including mixtures with lithium concentrations as low as 0.06 wt%. 66 The system operates at reduced temperatures (as low as 240 °C). Electrochemical tests demonstrated coulombic efficiencies up to 96% and lithium recovery rates exceeding 80%, with the purity exceeding 99.7 wt%.
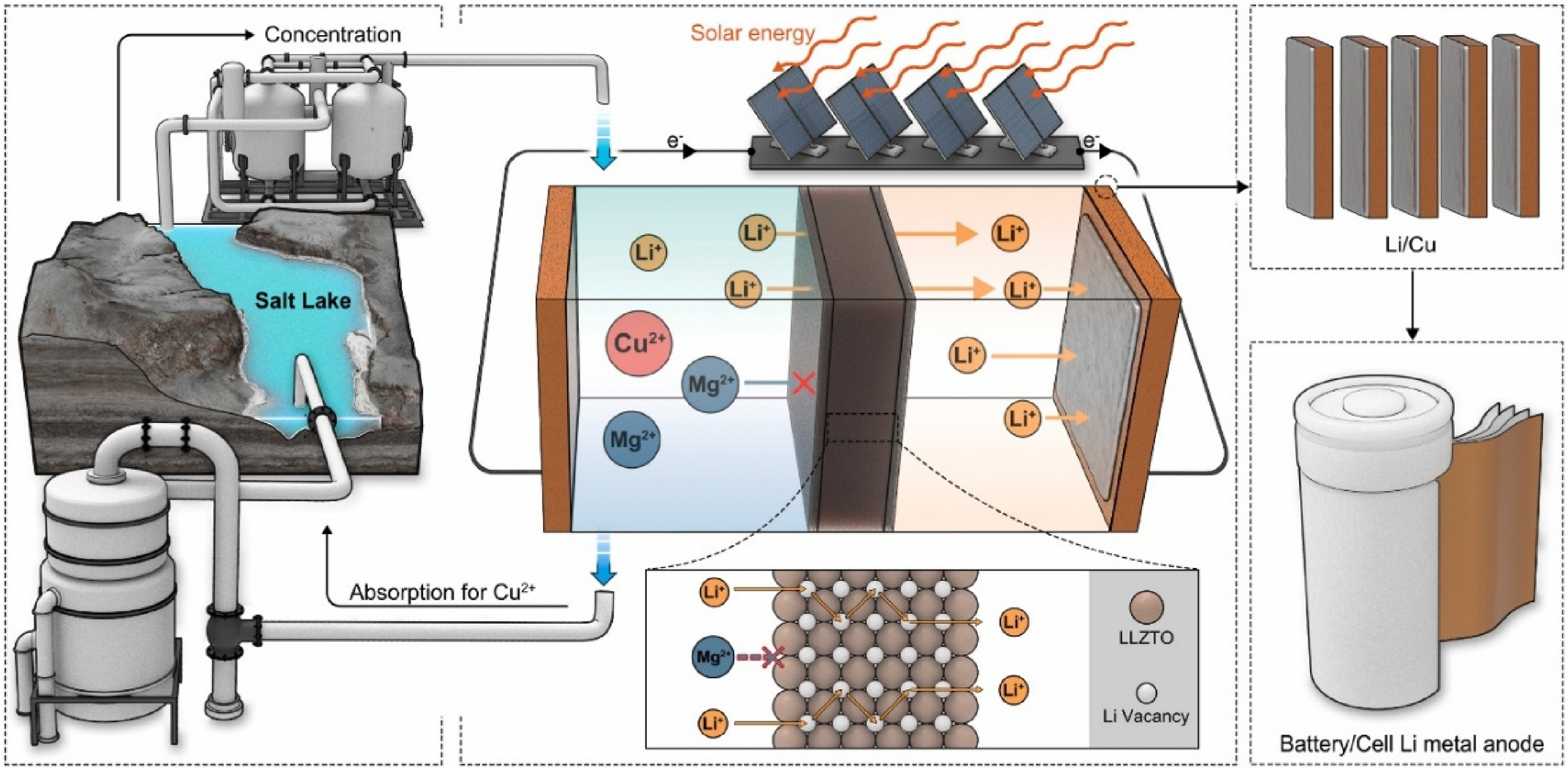
Schematic illustration of the DEEL system with a Li-free configuration and a sacrificial anode. Reproduced from Ref. 65
Furthermore, beyond primary extraction, innovative electrochemical methods have been proposed for direct lithium recovery from secondary sources. Fan et al. introduced an innovative electrochemical overcharging method to recover lithium metal directly from the cathodes of spent LIBs. 63 An overpotential of 5 V was applied to cathode materials such as LiCoO2 and LiFePO4, which can extract up to 90% of lithium metal. The lithium was then deposited on the copper substrate as thin foils with a thickness of around 40 µm with spherical morphology (Figure 14). This ultrathin lithium metal is particularly beneficial for enhancing the cycle life of LMBs, especially when used as a prelithiation material. Furthermore, this method is estimated to significantly reduce energy consumption to just $1.4/kg-Li and increase the product value by $79.1/kg-Li compared to traditional recycling approaches, as shown in Figure 15.

SEM image of deposited lithium metal. Reproduced from Ref. 63
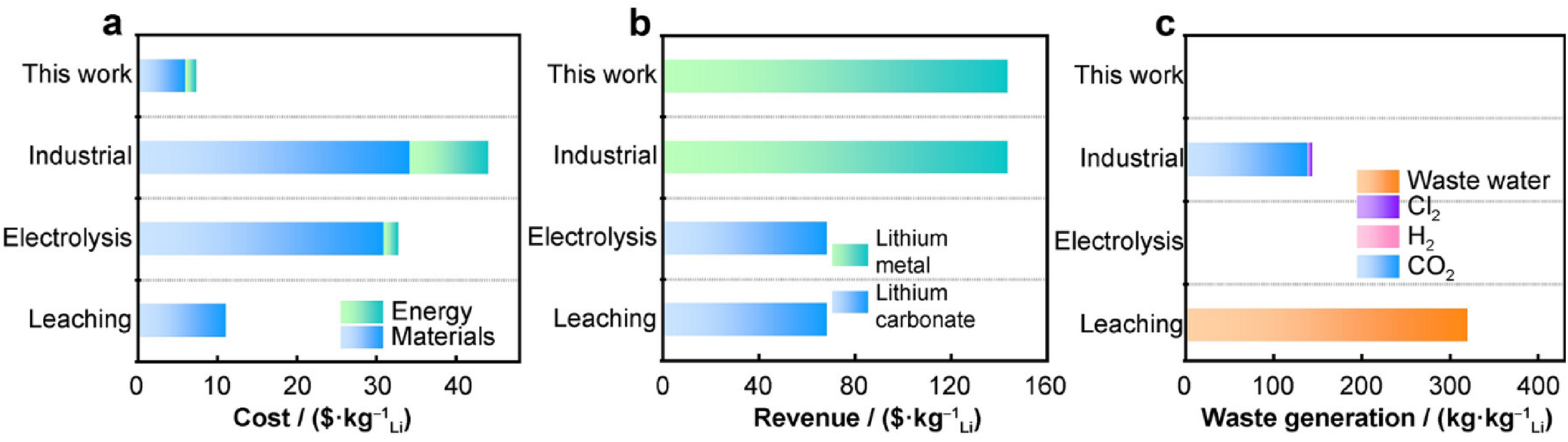
Economical and environmental analysis of four lithium recycling methods: a cost; b revenue; c waste generation. Reproduced from Ref. 63
Differently, Yang et al. demonstrated a solar-powered photoelectrochemical (PEC) system for selective extraction of high-purity lithium from multi-cation electrolytes derived from waste batteries. 73 The system uses a coplanar Si-based photocathode coupled with a TiO2 photoanode, operating at a low applied potential of 2 V under 1 sun illumination, exploiting redox potential differences between Li and other metal ions (Fe, Co, Ni). It achieved a lithium production rate of ∼1.38 g/hm−2 with 90.7% Faradaic efficiency and 99.5% purity, showing stable performance over multiple cycles and economic viability for lithium recycling.
Overall, the diverse innovative electrochemical approaches highlight significant progress toward addressing the limitations of conventional lithium metal production. Hybrid systems, membrane-assisted electrolysis, and solid-state electrolytes have collectively improved energy efficiency, selectivity, and product purity, while also enabling lithium extraction from complex resources such as seawater, brines, and recycled battery materials. In particular, solid-state and membrane-based systems offer clear advantages in suppressing parasitic reactions and achieving high coulombic efficiencies (>95%) with excellent purity. Meanwhile, emerging strategies such as photoelectrochemical and solar-assisted systems introduce alternative low-energy pathways that align with the increasing demand for sustainable and low-carbon lithium production.
However, despite these promising developments, several critical challenges remain unresolved, particularly regarding scalability, process stability, and economic competitiveness. Many of these systems rely on advanced materials (e.g., LLZO, LLZTO, LAGP) that are costly and difficult to manufacture at large scale, while also suffering from interfacial resistance, mechanical fragility, or long-term degradation. Although strategies such as interfacial engineering (e.g., TiO2 interlayers) and defect-tolerant structures have shown improvements, these solutions often add further complexity to system design. In addition, while membrane-based and solid-state approaches effectively address selectivity and purity, they are typically constrained by low current densities and lithium flux, which limit production rates and thus industrial throughput. Similarly, hybrid and flow-based electrochemical systems, although more scalable in principle, still operate at elevated temperatures or involve design complexities (e.g., pressure control, freeze-seal mechanisms) that may offset their efficiency gains.
Consequently, while next-generation electrochemical lithium production technologies demonstrate clear technical potential, their industrial viability remains contingent on achieving a balance between performance, cost, and scalability. Future progress will likely depend on the development of low-cost, robust electrolyte materials, improved interfacial engineering to enable higher current densities, and simplified system architectures that reduce operational complexity.
Metallothermic reduction of Li compounds
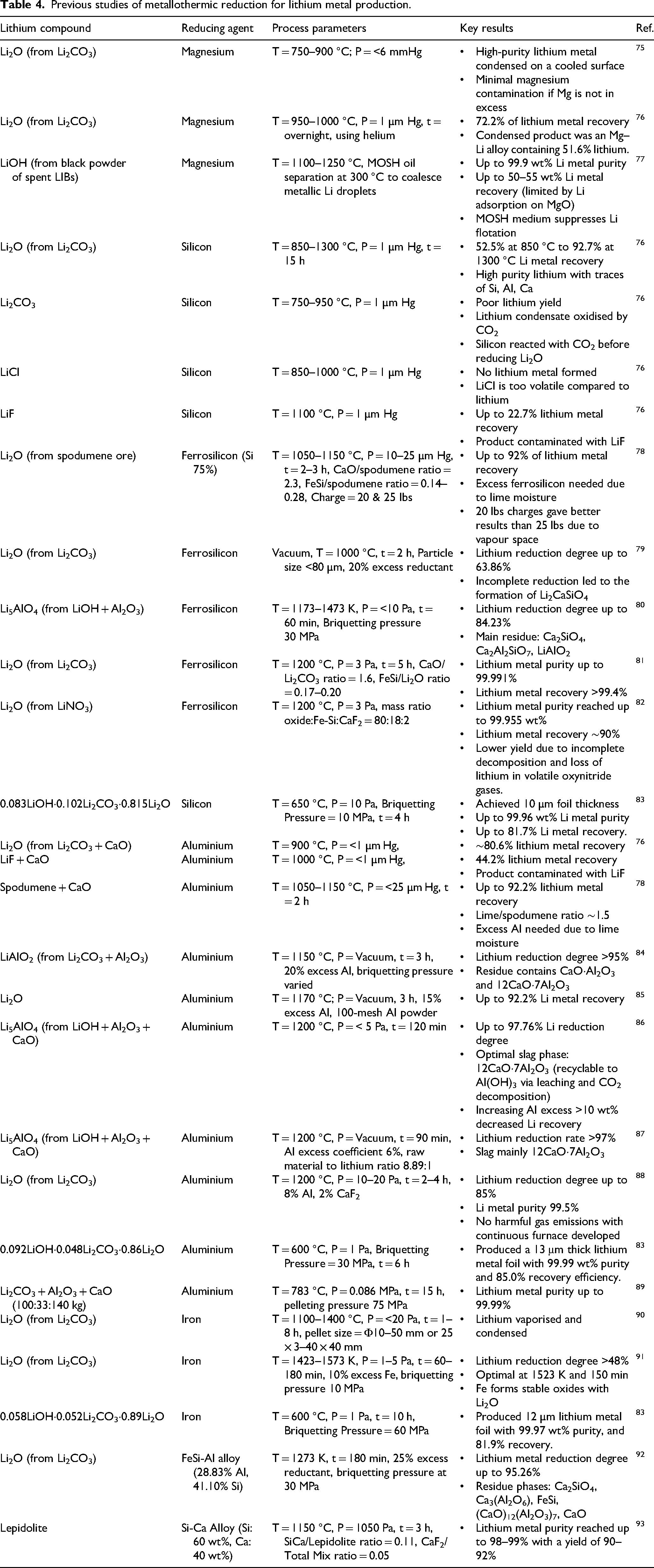
The utilisation of metallothermic reactions to reduce metal oxides has been widely explored due to their potential and advantages. Unlike carbothermic reduction, the use of metals as reducing agents does not produce environmentally harmful gases such as CO or CO2. Although metals are generally more expensive than carbonaceous materials, they offer the possibility of being recovered and reused through recycling or purification using additional treatment processes. 74 In the context of lithium metal production, Table 4 shows previous studies that used various metals as reducing agents, including aluminium, magnesium, and silicon (and ferrosilicon).
Previous studies of metallothermic reduction for lithium metal production.
Magnesium as reductant
Magnesium has been one of the earliest metals employed in the metallothermic reduction of lithium-bearing compounds, initially explored in early patents for reducing Li2O as the primary lithium source. In a patent from Hanson, the reduction process was carried out in a vacuum retort (below 6 mmHg) equipped with a water-cooled condenser and operated at 750–900 °C. 75 A controlled pellet injection technique was applied, introducing pellets one at a time into the hot zone of the retort to maintain low pressure and maximise lithium recovery. Each pellet reacted rapidly, releasing lithium vapour and gaseous by-products, which were continuously removed by a vacuum pump to prevent pressure build-up. The lithium vapour then condensed on the cooled upper section of the retort. Furthermore, it was found that the reaction rate was significantly influenced by feedstock grain (particle) size. The feedstock ground to 100 mesh (149 µm) resulted in a highly rapid and even explosive reaction, whereas 25 mesh (707 µm) particles produced a slower reaction that occurred over several minutes. However, the process often yielded a lithium-magnesium alloy rather than pure lithium metal. This is due to the volatility of magnesium during the reaction and the difficulty in maintaining ideal low-pressure conditions required for lithium vaporisation.
In another study, Kroll and Schlechten conducted magnesium reduction of Li2O under a high vacuum of less than 1 mmHg. 76 Different from the previous approach, this study utilised a batch-type vacuum furnace equipped with a porcelain reaction tube, oil-diffusion pump, and stainless-steel condensate trap, allowing for extended high-temperature exposure and precise control of pressure and temperature. The results demonstrated that only 72.2% of lithium could be recovered, while the purity reached 51.6%, with the remainder being magnesium. Despite successful reduction, the presence of magnesium in the condensate compromised the purity of the lithium product, making it difficult to separate lithium from magnesium due to their similar volatilities.
To address this intrinsic limitation of Mg-based systems, Joo et al. proposed a combustion–magnesiothermic reduction process combined with a post-reaction separation step for lithium recovery from spent battery black powder 77 as shown in Figure 16. LiOH was first prepared as an intermediate and then reduced with Mg via a self-sustaining exothermic reaction, producing a Li–Mg metallic phase within an MgO matrix rather than free lithium. To minimise Mg contamination, a high-temperature separation step using mineral oil saturated hydrocarbons (MOSH) at ∼300 °C was applied to selectively extract lithium from the Mg-containing phase. This integrated approach yielded lithium with reported purity up to 99.9%, although the overall recovery remained moderate (∼50–55%).
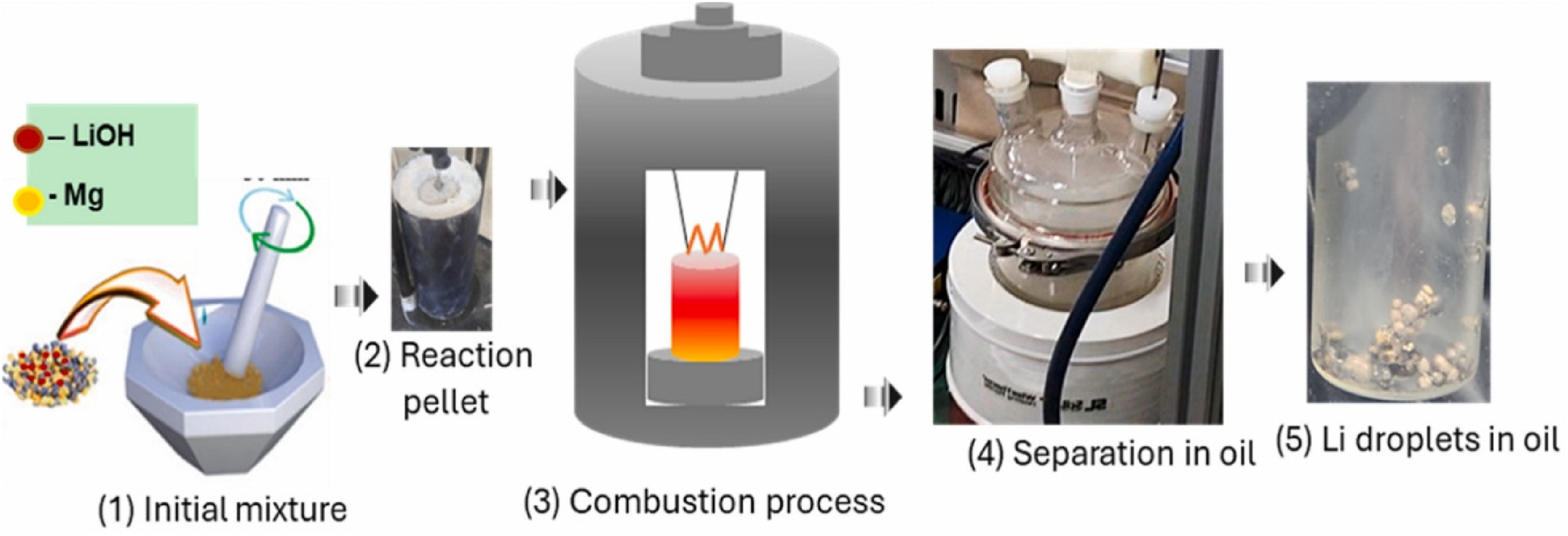
The schematics demonstrate the recycling of LiOH into Li metal. Reproduced from Ref. 77
Silicon as reductant
Silicon is a viable reducing agent for producing lithium metal. Besides its pure metallic form, silicon can also be used in the form of ferrosilicon. Kroll and Schlechten utilised 100-mesh metallic silicon to reduce Li2O. 76 The mixture was prepared with a 10% excess of silicon relative to the stoichiometric requirement. The reduction process was conducted under a vacuum pressure of 1 mmHg, with temperatures ranging from 850 °C to 1300 °C. The results demonstrated that increasing the temperature from 850 °C to 1300 °C significantly improved the reduction efficiency, from 52.5% to 92.7%, with purity reaching up to 99.94%. Thus, alternative lithium compounds such as Li2CO3, LiCl, and LiF were also evaluated as feedstocks. Lithium carbonate was found to decompose into Li2O before reduction, requiring a two-step process: initial heating at 750 °C to remove CO2, followed by reduction at 950 °C. However, the lithium condensate exhibited signs of oxidation, likely due to the presence of carbon dioxide released during the decomposition process. The recovery rate was notably low, attributed to the reaction between silicon and CO2, which inhibited the formation of Li2O and consequently its reduction. In contrast, the reduction of lithium chloride with silicon at 850 °C and 1000 °C resulted in no lithium metal formation, due to the high volatility of lithium chloride. Meanwhile, the reduction of lithium fluoride with silicon at 1100 °C achieved a modest lithium recovery of only 22.7%.
In the same year, Stauffer reported lithium metal production from spodumene ore through a metallothermic reduction process using ferrosilicon (75% Si) as the reductant. 78 The ore was mixed with CaO, and the reduction was then carried out under vacuum (25 µm Hg) at 1050–1150 °C for 2 h. A CaO-to-spodumene ratio of 2.3 and an excess ferrosilicon ratio of 0.14 yielded up to 87% lithium. Furthermore, a pilot-scale test was also conducted using a charge composition of 28.8% spodumene, 3.9% ferrosilicon, and 67.3% lime, achieving 85% lithium recovery at 1100 °C for 3 h. However, it was found that CaO absorbed moisture over time, resulting in a decrease in performance. To compensate for losses due to reactions with moisture in CaO, the ferrosilicon content was increased to 7.8%, while reducing the spodumene and CaO additions to 27.7% and 64.5% lime, respectively. With this adjustment, recovery reached 91% at 1150 °C using a 20 lbs charge over 2–2.5 h. A 20 lbs charge is considered the optimum weight, as increasing the charge to 25 lbs reduced the yield to 69%, possibly due to restricted vapour escape, highlighting the importance of charge volume and packing in maximising lithium recovery.
A study from Yuezhong et al. demonstrated the reduction of Li5AlO4 clinker (prepared from LiOH and Al2O3) with ferrosilicon (equations (9) and (10)).
80
The use of Li5AlO4 clinker could offer an effective alternative to Li2CO3, which requires excessive CaO and produces large amounts of slag. The reduction process was carried out at various temperatures (1173–1473 K) for a constant duration of 60 min. A strong dependence of the lithium reduction degree on temperature was observed, as the reduction degree was only 35.58% at 1173 K, but it increased significantly to 84.23% at 1473 K as shown in Figure 17(a). The residue from the reduction process mainly consists of Ca2SiO4, Ca2Al2SiO7, CaO and LiAlO2 (Figure 17(b)).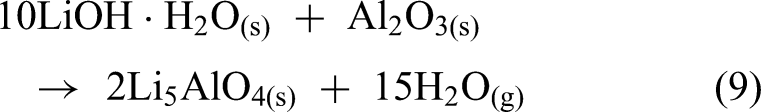
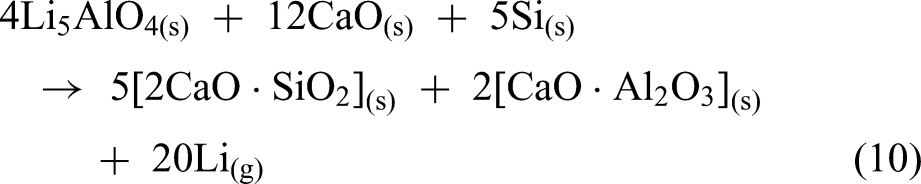
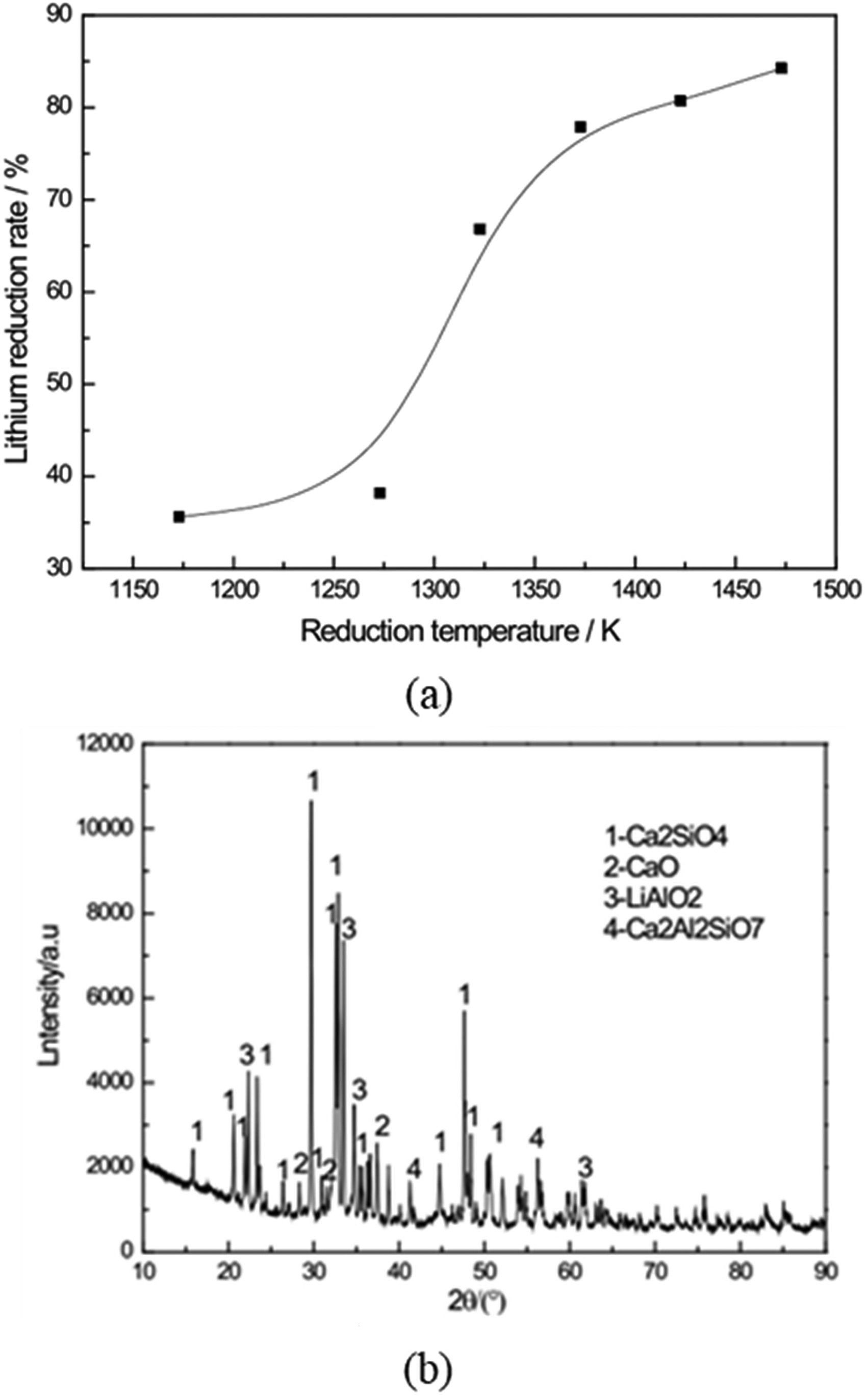
(a) Lithium reduction degree at different reduction temperatures (b) XRD of reduction residue at 1473 K. Reproduced from Ref. 80
Visual observations in Figure 18(a) supported this trend, where briquettes with low reduction degrees remained solid. Meanwhile, those with higher degrees became powdery, indicating structural breakdown due to the reduction reaction. Despite the high reduction efficiency at elevated temperatures, the phase analysis of residues revealed the appearance of LiAlO2, suggesting that the reduction was not fully complete. Additionally, a white powdery “crystallised product” was observed in the crystalliser (Figure 18(b)); however, its exact composition and crystalline phase were not determined.

(a) Visual observation of reduction residues at different reduction temperatures (b) reduction product in the crystalliser. Reproduced from Ref. 80
Aluminium as reductant
Aluminium is an effective reductant for lithium-bearing compounds due to its strong affinity for oxygen, which drives the reduction of lithium compounds and the formation of stable alumina-based slags. This characteristic facilitates lithium metal extraction by promoting the release of lithium vapour during metallothermic reduction. In one study, Yuezhong et al. investigated the reduction of LiAlO2 pellets, synthesised from Li2CO3 and Al2O3, with the addition of CaO (molar ratio 30:30:40%, respectively). 84 Reduction degree increased significantly with temperature, from 40.2% at 900 °C to over 93% at 1150 °C after 2 h with 10% excess aluminium. Extending the reduction time to 3 h or increasing the excess aluminium to 20% resulted in only slight improvements, while changes in particle size (from 80 to 75 µm) had a negligible impact. Residual phases confirmed effective binding of alumina by CaO, reducing lithium aluminate formation.
Building on a previous study, the same group expanded the exploration to Li5AlO4 synthesised from LiOH, CaO, and Al2O3 in a molar ratio of 70:7:32, with the expected reaction as shown in equation (11).
87
Higher temperatures generally improved the reduction degree, although the increment from 1100 to 1200 °C was minimal when the holding time was constant at 60–90 min. Reaction time showed a stronger effect, with the reduction degree at 1200 °C increasing by over 30% from 30 to 60 min and reaching 90% at 75 min, before plateauing beyond 90 min. As shown in Figure 19(a), adding excess aluminium up to 6% increased the reduction degree to 97%, with no trace of lithium compound in the residue (Figure 19(b)). However, as shown in Figure 19(c), additional aluminium up to 20% decreased the reduction efficiency, as evidenced by the formation of LiAlO2 in the residue. Morphological comparison of slags from 6% and 20% excess aluminium further shows that excessive aluminium forms a coating layer (Figure 20(a) and (b)). This coating layer likely acts as a diffusion barrier that restricts mass transfer and inhibits continuous contact between the reactants, thereby suppressing further lithium reduction and volatilisation.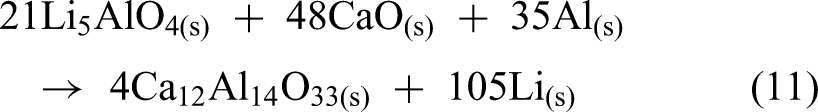
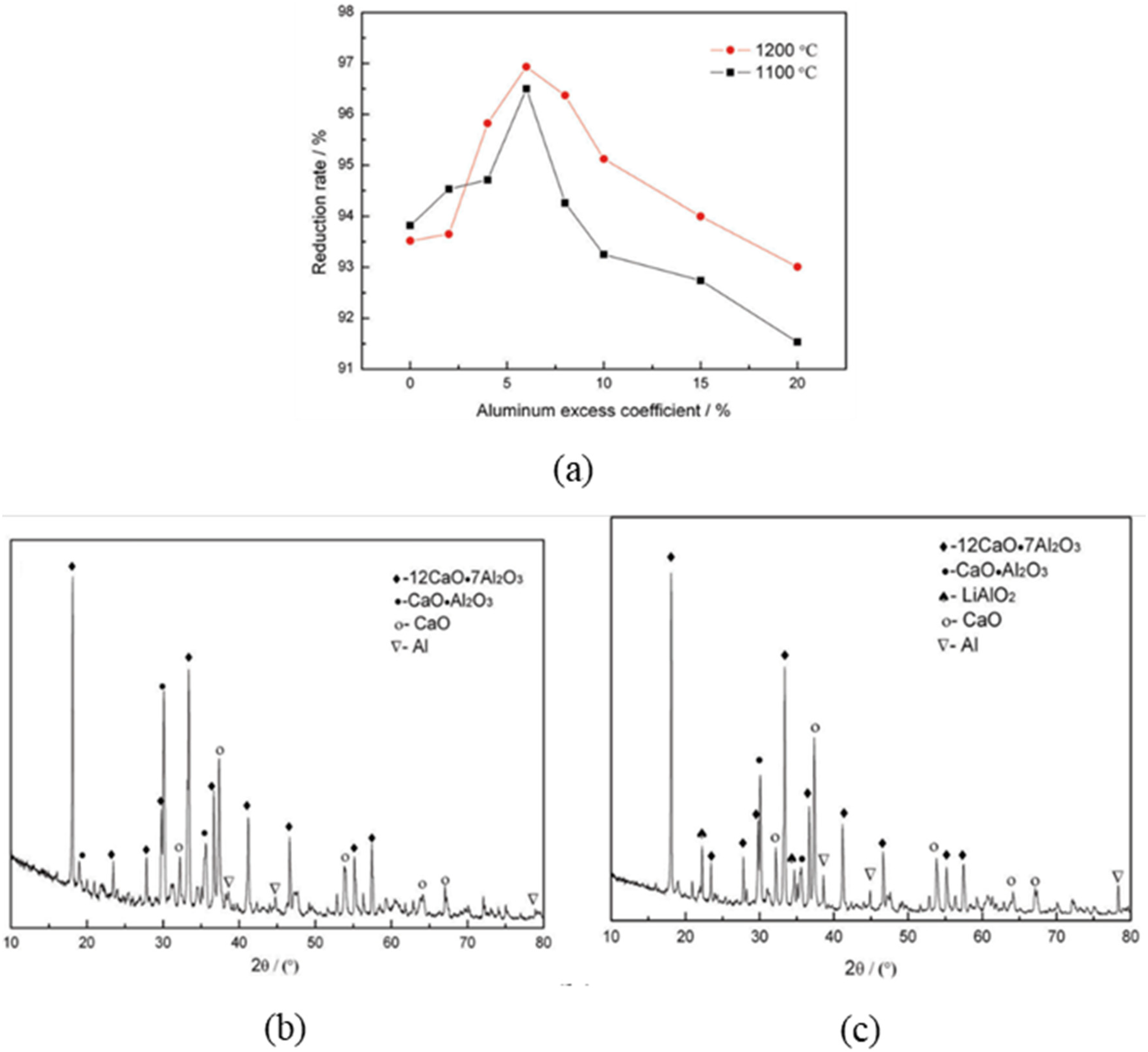
(a) Lithium reduction degree as functions of the aluminium excess coefficient (reduction temperature 1100 °C and 1200 °C, reduction time 90 min) (b) XRD patterns of the reduction slag obtained using an aluminium excess coefficient of 6% and (c) 20%. Reproduced from Ref. 87

(a) SEM of residue with temperature 1200 °C, time 90 min, Al excess coefficient 6%, and (b) SEM of residue with temperature 1200 °C, time 90 min, Al excess coefficient 20%. Reproduced from Ref. 87
On the other hand, Lu and Neelameggham applied aluminothermic reduction using Li2CO3 and CaO (mass ratio 1:2) to prevent melting during the decomposition of Li2CO3. 88 The mixture was then combined with 8% Al and 2% CaF2 before pelletising. The reduction process was carried out in a 120-kW furnace and yielded lithium ingots with 99.5% purity (main impurity sodium), 85% lithium recovery, and 90% current efficiency. Slag composition confirmed the important role of CaO in binding alumina. Later, they found that an excessive amount of Al can hinder the reduction process and using a smaller amount of Al can improve the recovery up to 97.76%. 94
A recent study from Nababan et al. demonstrated that thermodynamic modelling of lithium cobalt oxide (LCO), a widely used cathode material in lithium-ion batteries, with aluminium predicted lithium vaporisation can occur at temperatures as low as 840–920 °C at low pressure (∼10−5 atm) compared to >1700 °C under inert atmospheres. Moreover, experimental results confirmed that the highly exothermic thermite reaction raises the system temperature beyond the melting point, allowing lithium to volatilise and later condense as metallic lithium on the condenser, with the possible mechanism as shown in Figure 21. Although some lithium was recovered as LiOH due to post-reaction hydration, the process shows clear potential for direct lithium metal production if condensation and handling are optimised. 95
Conceptual diagram of the aluminothermic reduction pathway for LiCoO2 with CaO flux. Reproduced from Ref. 95
Iron as reductant
The development of lithium production through metallothermic reduction has gained significant attention in recent years. However, most studies utilise metals such as Al, Mg, or Si as reducing agents, which are relatively expensive. In response to this, Fe has emerged as a potential reducing agent due to its advantages, including low cost, abundant availability, and ease of procurement. From a thermodynamic standpoint, using Fe to reduce Li2O under atmospheric pressure is not feasible due to the unfavourable Gibbs free energy of the reaction. However, under vacuum conditions, the continuous removal of gaseous lithium metal shifts the equilibrium, making the reduction thermodynamically viable. This approach enables the reaction to proceed by suppressing the reverse formation of Li2O, thereby overcoming the limitations of ambient-pressure systems.
This approach was investigated by a research group from Kunming University of Science and Technology, who developed the iron-based vacuum reduction method by specifying a two-step thermal treatment with defined temperature ranges (approximately 700 °C for calcination and 1100–1400 °C for reduction) and reaction times (1–8 h). 90 Moreover, they also emphasise process optimisation aspects such as heating rates, briquette dimensions, and vacuum conditions (up to 20 Pa). Later, the same group employed a water-cooled condenser installed at the top of the furnace to collect condensed lithium vapour. 91 Optimal reduction (about 60%) occurred at 1573 K, 2.5 h, 10% excess Fe, and 10 MPa briquetting pressure. Excess Fe above 10% lowered efficiency due to lithium–iron oxide phase formation, which can immobilise lithium in stable compounds and reduce its volatilisation during reduction. Meanwhile higher briquetting pressure hindered lithium vapour release by creating denser briquettes. Condensate analysis showed Li2O and LiOH due to lithium reaction with moisture and oxygen, while the slag phase contained lithium–iron oxides, unreacted Fe, and iron oxides.
Carbothermic reduction of Li compounds
Carbothermic reduction has emerged as an interesting alternative for lithium metal production, offering several advantages over industrial methods. The use of carbon as a reducing agent is desirable due to its low cost, availability, and relatively simple reaction mechanism. The potential of this method has been recognised as early as 1937, when the U.S. Bureau of Mines first attempted lithium metal production via the direct carbothermic reduction of spodumene. 96 However, the process yielded low lithium recovery and led to the formation of lithium carbide (Li2C2). Moreover, the reaction was conducted at extremely high temperatures (∼1680 °C), which not only posed challenges in energy efficiency and material stability but also contributed to the decline in interest for this route. These early limitations, nonetheless, have sparked further research aimed at optimising reaction conditions and exploring alternative lithium precursors to enhance yield and feasibility (Table 5).
Previous studies of carbothermic reduction for lithium metal production.

Based on these early efforts, recent studies have reconsidered carbothermic reduction for producing lithium metal to improve reaction control and reduce impurities. Chen observed the reduction behaviour of Li2O using activated carbon as a reducing agent under vacuum conditions (20 Pa). 97 The Li2O used in the reduction process was obtained through thermal decomposition of Li2CO3. Before decomposition, CaO was added for fluxing, resulting in a final composition of Li2O feedstock containing 19.83 wt.% CaO. The reduction experiments were performed at a controlled pressure of 20 Pa, with temperatures set at 1373 K and 1423 K, and reduction times ranging from 0 to 120 min. It was noted that higher temperatures and longer durations significantly affected the reduction rate. However, the lithium purity was considerably low (54.34 wt.%), with carbon and oxygen being the major impurities (24.80% and 20.69%, respectively) in the product, suggesting reoxidation of lithium vapour by CO during condensation.
In other work, Lin et al. explored the potential of calcium carbide (CaC2) as a reducing agent to produce lithium metal using Li2CO3 as the precursor. 98 The process was conducted under a vacuum pressure of 5 Pa. The obtained lithium vapour was condensed using a water-cooled disk to obtain lithium metal. Maximum recovery of 92.69% was achieved at a temperature of 1373 K with reducing time of 80 min, but the lithium metal purity remained relatively low (58.28%). This reduction in purity was attributed to the dominant reducing role of calcium, which led to incomplete reactions and the presence of unreacted carbon. At elevated temperatures, carbon volatilisation further contributed to impurity formation by reacting with lithium vapour to form Li2C2. Furthermore, the condensed lithium metal appeared greyish-black and turned yellowish-brown upon exposure to air; however, no visual or spectral evidence was provided to validate this observation. They also revealed the presence of secondary phases in the condensate, such as LiOH·H2O and Li3N, indicating that additional purification is necessary.
To address the challenges related to precursor stability and reaction control, Shi et al. investigated lithium aluminate (LiAlO2) as a more thermally stable precursor for carbothermic reduction.
99
The possible reduction reactions between LiAlO2 and carbon can be expressed as shown in equations (12) to (14).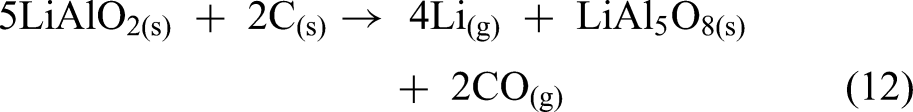
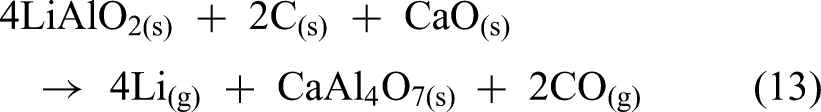
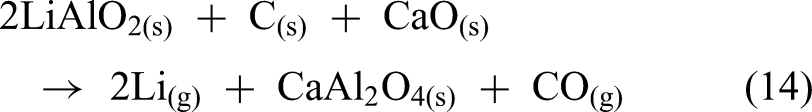
Under vacuum conditions (10 Pa), carbothermic reduction of LiAlO2 using coking coal and CaO as an additive demonstrated increasing reduction degree with higher temperature, reaching a maximum of 79% at 1623 K. A reduction time of 2 h was found to be sufficient for reaction completion, as extending the duration to 5 h resulted in negligible improvement. The optimal carbon-to-LiAlO2 molar ratio was determined to be 1.0. Notably, the addition of 20 wt.% CaO significantly enhanced lithium recovery to nearly 100%, which was attributed to the suppression of LiAl5O8 formation as shown in Figure 22(a) and (b). However, the mechanism for this suppression was not explained. Despite these promising results, a critical limitation of the study remains in the absence of any lithium vapour capture or condensation strategy. No apparatus was described for isolating or collecting lithium metal, and the recovery conclusions were inferred solely from the depletion of lithium content in the slag phase.
(a) Relationship between different CaO additions and lithium recovery (b) XRD pattern of reducing residues at different additions of CaO. Reproduced from Ref. 99
Expanding beyond synthetic precursors, Lyu et al. examined carbothermic reduction of natural spodumene ore, which contains lithium alongside other valuable elements such as silicon and aluminium.
100
Spodumene was reduced using anthracite coal, with Fe2O3 added as a ferrosilicon-promoting agent to enable the simultaneous recovery of lithium, alumina (Al2O3), and silicon under a vacuum pressure of 20 Pa, as demonstrated by equation (15).
Notably, as displayed in Figure 23(a), increasing the temperature above 1648 K did not give a significant increase in the lithium reduction degree. In addition, the increasing temperature led to the appearance of CaAl12O19 (Figure 23(b)), indicating that both temperature and duration influence not only alumina stability but also overall process control. Later, Du et al. investigated the effect of adding Fe2O3 with a fixed composition ratio of spodumene, iron oxide powder, and anthracite at 5:5:3, respectively. 101 The operation was conducted under a lower pressure (10 Pa). However, the results showed that lithium removal above 95% could only be achieved at a temperature of 1823 K. Furthermore, the optimal reduction time was found to be 8 h, and extending the duration beyond this point did not produce any significant effect. This phenomenon indicates that the addition of the additive slows down the lithium extraction process, which may be attributed to the reaction between carbon and the additive (in this case, Fe2O3) at lower temperatures.
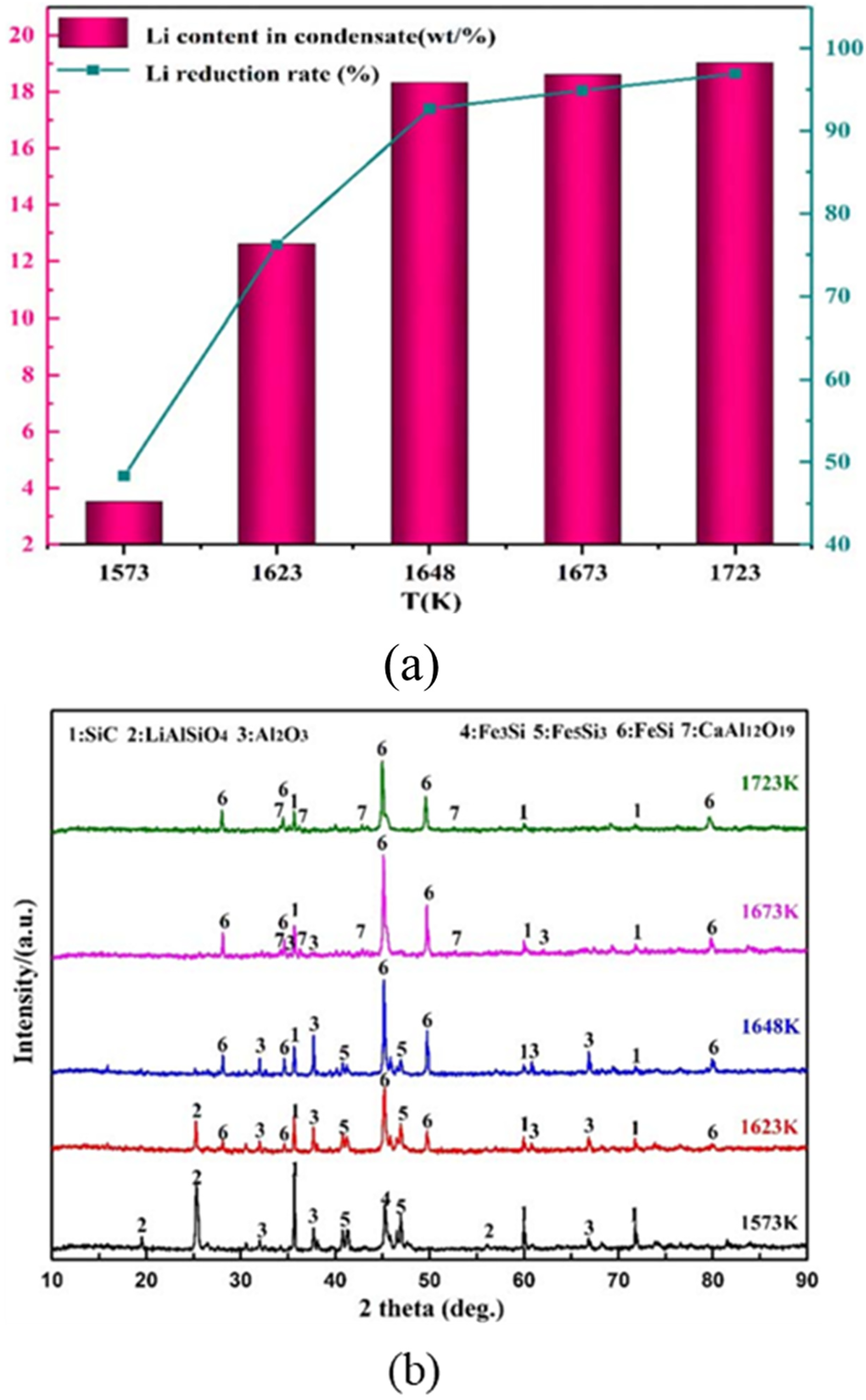
(a) Effects of temperature on Li reduction degree and Li content-in-condensate (b) XRD pattern of the residues. Reproduced from Ref. 100
Despite the promising outcomes of recent studies, a critical weakness lies in the minimal explanation of the lithium vapour capture strategy. In the study by Shi et al., no system for condensing or isolating lithium metal was described (Figure 24), and conclusions were drawn solely from lithium depletion in the slag. Meanwhile, Chen et al., 97 Lin et al., 98 and Lyu et al. 100 (Figure 25) incorporated a condensation unit to collect lithium vapour; however, their work lacked a clear explanation of the condensation mechanism, visual confirmation of lithium metal formation, and evaluation of the system's effectiveness or scalability. Du et al. later confirmed that capturing lithium vapour is challenging due to limited furnace airtightness, causing part of the vapour to escape or condense elsewhere in the furnace instead of on the condensation plate. 101 Therefore, these studies highlight a critical gap in lithium vapour handling and recovery, which is critical before its practical and scalable implementation.
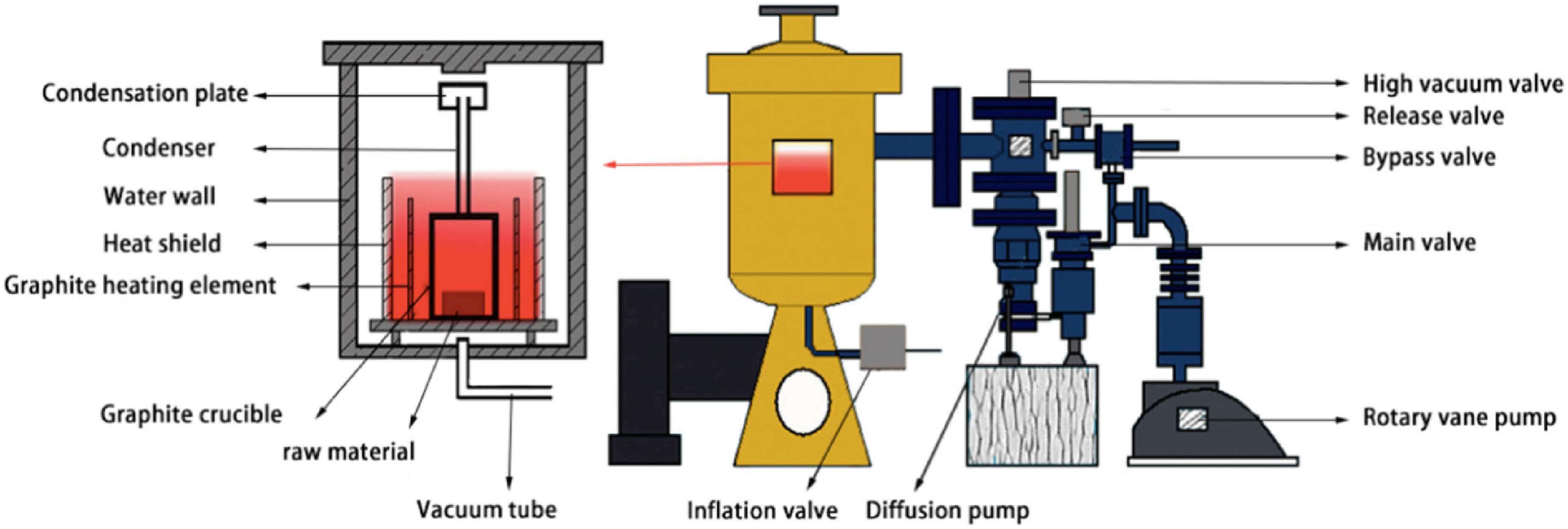
Schematic of a vacuum lithium reduction furnace: 1: reaction zone (crucible); 2: high-temperature graphite heating-element zone; 3: high-temperature graphite electrode zone; 4: high-temperature thermocouple zone; 5: graphite connection tube; 6: insulation shell; 7: thermal insulation cotton; 8: low-temperature thermoelectric zone; 9: low-temperature graphite electrode zone; 10: low-temperature graphite crucible zone; 11: low-temperature heating-element zone; 12: water-cooling jacket; and 13: vacuum pump interface. Reproduced from Ref. 99
Schematic diagram of the vacuum melting furnace. Reproduced from Ref. 100
To overcome this, CSIRO has developed the LithSonic technology, representing a novel approach to lithium vapour handling and metal recovery. 102 Adapted from a process originally developed for magnesium metal production, 103 LithSonic utilises a high-temperature carbothermic reaction exceeding 1600 °C at atmospheric pressure to generate lithium vapour. This vapour is then rapidly accelerated through a specially designed de Laval nozzle, achieving supersonic velocities before undergoing extremely rapid quenching at rates exceeding one million degrees per second. This supersonic quenching forces the condensation of lithium vapour into a solid phase under non-equilibrium conditions, hence effectively suppressing re-oxidation. Although still in the developmental stage, this approach shows an exciting potential in lithium production technologies.
Process-structure-performance relationships in lithium metal
Lithium metal production routes exhibit different process–structure–performance relationships that can be systematically compared through key operating parameters, including temperature, current density (where applicable), and transport pathway. Across technologies, these parameters control whether lithium formation is kinetically controlled or transport-limited, which in turn determines deposit morphology, impurity incorporation, and electrochemical performance when applied for LMBs.
From a process perspective, lithium metal production spans a wide operating window, ranging from low-temperature aqueous systems (15–95 °C), to industrial LiCl-KCl electrolysis (450–550 °C), and thermochemical reduction routes (>1000 °C). This variation is dictated by the chemical state of lithium in the feedstock and the stability of the processing medium. In molten salt LiCl–KCl systems, lithium exists as Li+ within a highly ionically conductive molten electrolyte but requires elevated temperatures to maintain the salt in a molten state and sustain sufficient ionic mobility. This high-temperature molten salt system provides ionic conductivity in the range of ∼1.5–2 S/cm at 450–550 °C 104 and Li+ diffusion coefficients on the order of 10−5–10−4 cm2/s. In contrast, low-temperature aqueous electrolysis-based systems, including aqueous leachates, brine-derived electrolytes, and hybrid aqueous–organic configurations, are characterised by the thermodynamic stability of these species in solution, hence, eliminates the need for high-temperature phase transition, allowing operation at 15–95 °C. However, the strong solvation environment and low thermal energy significantly reduce Li+ mobility, resulting in more severe mass transport constraints.
As a result, molten salt systems can sustain industrial current densities of approximately 0.3–1 A/cm2, whereas solution-based systems are generally limited to below ∼50 mA/cm2 due to earlier onset of transport limitations and parasitic reactions. These differences in achievable operating current density significantly influence the balance between reaction kinetics and mass transport during lithium deposition. Higher current density in molten salt electrolysis operation enables substantially greater lithium production rates but simultaneously increases susceptibility to transport limitations. As current density approaches ∼1 A/cm2, local Li+ depletion near the electrode surface becomes increasingly significant, driving the system toward diffusion-limited mechanism characterised by concentration gradients and morphological instability. In-situ observations by Natsui et al. 105 using high-speed optical microscopy revealed that lithium initially nucleates as liquid droplets on the electrode surface, followed by heterogeneous growth under elevated current conditions. As Li+ depletion becomes more severe, growth becomes increasingly non-uniform, leading to rough deposits and fragmentation into fine lithium droplets dispersed within the molten salt.
Differently, lower current densities in aqueous lithium electrochemical systems reduce deposition flux and concentration polarisation due to slower growth kinetics, promoting more uniform nucleation and finer lithium structures under the stronger influence of interfacial energy effects. Lithium morphology is governed by the competition between electrochemical overpotential, nucleation barrier, and interfacial energy at the Li/electrolyte interface. Compared to molten salt systems with more uniform interfacial conditions and reduced solvation effects, aqueous/organic electrolytes exhibit stronger Li+ solvation and higher nucleation barriers, promoting localised anisotropic growth and morphology evolution with current density. For instance, Mashtalir et al. 53 demonstrated that increasing current density can systematically tune lithium morphology from nanorods (3 mA/cm2) to spheres (5 mA/cm2) and cubes (7 mA/cm2), highlighting the strong coupling between current density and deposit morphology in low-temperature systems. Similarly, controlled aqueous electrolysis approaches have shown that lithium morphology is highly sensitive to operating conditions such as current density and deposition time, where prolonged electrolysis favours nanorod-like deposits while shorter deposition leads to denser spherical morphologies in lithium films. 55
At the extreme end, thermochemical reduction (>1000 °C) is required when lithium is bound in highly stable solid phases such as oxides and silicates. In these systems, lithium is not present as a mobile ionic species but is embedded within high lattice-energy structures, necessitating substantial thermal input to overcome both thermodynamic stability and solid-state diffusion barriers. Lithium metal formation is driven by high-temperature reduction reactions (e.g., carbothermic or metallothermic), governed by favourable Gibbs free energy at elevated temperatures rather than electrochemical potential. However, unlike electrochemical routes, these processes remain poorly characterised in terms of lithium morphology. Most studies focus on phase equilibria and conversion efficiency, while the physical form of the produced lithium is rarely reported. This gap arises from experimental challenges associated with extreme temperatures and the high reactivity of lithium, which hinder direct observation and preservation of the metallic phase.
Although lithium produced from these routes is not directly deployed as LMBs anodes, the as-produced characteristics, particularly impurity content, morphology, and microstructure, define the baseline material quality from which all downstream processing must proceed. These intrinsic features impose constraints on achievable purity, structural uniformity, and interfacial stability, thereby influencing the complexity and effectiveness of subsequent steps such as melting, casting, and rolling. For instance, lithium obtained from molten salt electrolysis, while enabling high production rates, may require additional purification and morphological control to mitigate heterogeneity introduced under transport-limited conditions. In contrast, finer lithium structures produced in solution-based systems may reduce shaping requirements but introduce challenges related to surface reactivity and stability. Consequently, the process-dependent structure of as-produced lithium not only reflects the governing formation mechanisms but also determines the processing pathways and practical limits for achieving reliable electrochemical performance in LMBs systems.
Challenges and future outlook
The rapid expansion of lithium-based energy storage technologies has intensified demand for scalable lithium metal production, while simultaneously elevating the importance of environmental and energy efficiency constraints in process selection. A meaningful assessment therefore must be based on direct cross-system comparison using unified performance indicators, including specific energy consumption, operating temperature, current efficiency, product purity, and technology readiness level (TRL) (Table 6). Although data availability varies across routes, comparative trends can still be identified and used to define key bottlenecks and future research priorities.
Key metrics comparison of each lithium metal production route.

From an energy and process severity perspective, a clear stratification exists between aqueous, molten, and high-temperature thermochemical systems. Aqueous electrolysis exhibits the lowest reported energy demand (8–20 kWh/kg Li) and operates at near-ambient conditions (15–95 °C) but remains limited by very low TRL (1–3) and incomplete process validation at scale. In addition, these systems typically involve multiple chemical processing steps associated with lithium extraction and purification, which may introduce additional environmental burdens compared to other routes. In contrast, industrial molten salt electrolysis requires significantly higher thermal input (400–500 °C) but operates within a relatively narrow and industrially stabilised energy window (30–40 kWh/kg Li), representing the only fully commercialised route (TRL 9). Thermochemical routes occupy the highest temperature mode (600–1680 °C), indicating a shift from electrical to thermal energy dominance; however, the absence of reliable system-level energy consumption data for these processes represents a critical gap in the field, preventing rigorous benchmarking against electrochemical technologies.
A different trade-off emerges when process efficiency and product quality are considered. Molten carbonate electrolysis demonstrates the highest reported current efficiency (90–97%) while simultaneously achieving high product purity (99–99.5%), approaching or even exceeding the performance of industrial LiCl–KCl electrolysis (80–90% efficiency, 98–99% purity). Solid-state and non-aqueous electrolysis systems also exhibit high efficiency (∼97%) and comparable purity (99.5–99.7%) but remain at early TRL (1–3), limiting their industrial relevance. In contrast, carbothermic reduction shows the widest variability in efficiency (54–95%), reflecting strong sensitivity to reaction conditions and incomplete control over lithium vapour formation, transport, and recovery. Collectively, these observations indicate that many emerging electrochemical systems are already approaching parity with industrial processes in intrinsic performance, and that the primary barrier to their adoption is no longer efficiency, but engineering scalability and system integration.
When TRL is considered alongside performance metrics, a clear separation between maturity and performance potential becomes evident. Industrial LiCl–KCl electrolysis remains the only fully mature technology (TRL 9), despite not being the most energy-efficient or environmentally favourable option. All alternative routes cluster within TRL 1–5, indicating that the field is characterised more by laboratory-scale diversity than by industrial convergence. This mismatch demonstrates that the dominant barrier across emerging technologies is scale-up engineering rather than fundamental chemistry, suggesting that future progress will depend more on advances in reactor design, materials durability, and process integration than on the discovery of new reaction pathways.
From a system-level perspective, three distinct technological groups can be identified. The first group consists of mature but moderately efficient systems (industrial molten salt electrolysis), characterised by stable operation but constrained by environmental burdens and strict feedstock requirements. The second group includes high-efficiency but low-TRL electrochemical systems, such as molten carbonate, solid-state, and aqueous processes, which demonstrate strong electrochemical performance but lack industrial scalability. The third group comprises thermochemical routes, distinguished by extreme temperature requirements and broad feedstock flexibility, but limited by insufficient kinetic understanding and incomplete process controllability. This classification highlights that the field is not converging toward a single dominant solution but rather diverging into specialised technological pathways with distinct roles across the future lithium supply chain.
Based on the comparisons discussed above, several key perspectives can be identified to guide the future development of lithium metal production:
Future industrial electrolysis systems should move towards tolerating lower-grade LiCl feedstocks (<99 wt%), coupled with closed-loop Cl2 recycling systems. This would directly reduce both energy intensity and environmental burden. Additionally, exploring alternative electrolyte chemistries with lower melting points could enable operation below 400 °C, improving overall efficiency. Among emerging routes, carbothermic reduction shows the strongest long-term potential, particularly when coupled with biomass-derived carbon, which could enable near net-zero emissions. However, quantitative studies on reaction kinetics, heat integration, and lithium vapour yield are critically lacking and must be addressed to move beyond TRL 3–4. Regardless of thermochemical route, efficient lithium recovery depends on advanced vapour handling. Technologies such as controlled condensation (e.g., LithSonic-type approaches) should be systematically evaluated for capture efficiency, purity retention, and scalability. Future lithium production should not be viewed as a standalone process. Opportunities exist for CO utilisation in metallurgical systems (e.g., blast furnaces), recycling of process chemicals in electrochemical routes, integration with lithium recovery from seawater and spent LIBs
Conclusions
Lithium remains a critical metal with increasing demand, particularly due to the development of energy storage technologies, such as batteries. However, the electrolysis method, currently the primary technology for lithium production, still has many limitations, including the need for high-purity raw materials, process inefficiencies, and significant environmental impacts. Meanwhile, alternative routes, such as thermochemical and low-temperature electrochemical processes, also face significant challenges before they can be implemented on an industrial scale. Overall, the literature indicates that no existing method at present fully meets the criteria for sustainable, scalable, and low-emission lithium metal production.
Therefore, future research should prioritise improving electrolysis efficiency, developing greener carbothermic processes with the application of biomass and its derivatives as reducing agents, and developing more advanced lithium vapour handling technologies. Furthermore, exploring unconventional lithium sources such as brine, seawater, and recycled end-of-life batteries is crucial for establishing a more sustainable and efficient lithium metal supply chain. By highlighting the key challenges and opportunities, this review provides a foundation for guiding future advancements toward cleaner and more resilient lithium metal production.
Supplemental Material
sj-docx-1-inr-10.1177_09506608261464388 - Supplemental material for Advances in lithium metal production technologies
Supplemental material, sj-docx-1-inr-10.1177_09506608261464388 for Advances in lithium metal production technologies by M Bagas Ananda, Bintang A Nuraeni, Dongmei Liu, Warren Bruckard and M Akbar Rhamdhani in International Materials Reviews
Footnotes
List of symbols and acronyms
Acknowledgements
The author gratefully acknowledges the support from the Commonwealth Scientific and Industrial Research Organisation (CSIRO) and Swinburne University of Technology through the Growth Swinburne University Postgraduate Research Award (SUPRA) funding scheme.
Author contribution(s)
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.