
Abstract
This paper addresses some aspects of the analysis of cross-over trials with missing or incomplete data. A literature review on the topic reveals that many proposals provide correct results under the missing completely at random assumption while only some consider the more general missing at random situation. It is argued that mixed-effects models have a role in this context to recover some of the missing intra-subject from the inter-subject information, in particular when missingness is ignorable. Eventually, sensitivity analyses to deal with more general missingness mechanisms are presented.
1 Introduction
The issue of missing data in clinical trials is almost ubiquitous. While this is well recognized for adequate and well-controlled trials and to some extent addressed by regulatory guidelines, 1 it seems to be less recognized for trials in early drug development. During that development phase pharmacokinetic studies play a role as first in man, bioequivalence (BE), drug–drug interaction (DDI), or food interaction studies which frequently apply a cross-over design. The complete case (CC) analysis as proposed by Grizzle 2 that considers only subjects that completed the entire sequence of periods is often still considered the benchmark method despite its proneness for biased results.
An early reference on the analysis of incomplete data in a two-period cross-over design is Patel’s 3 paper. This paper suggests a maximum-likelihood estimator under the assumption of missingness in period 2 only. No restrictions on the variances of the response variable in different periods or under different treatments are made. Kenward and Molenberghs 4 pointed out that Patel’s method could not be applied in a missing at random (MAR) situation because his precision estimator is based on a missing completely at random (MCAR) framework. Lee et al. 5 modified the method to provide correct estimates under MAR. (A definition of the different missingness mechanisms are provided in Section 2.)
Some authors focused attention on three-period two-treatment cross-over designs to be able to estimate carry-over effects which is not possible in 2 × 2 cross-over studies. Richardson and Flack 6 studied maximum-likelihood estimators and imputation methods for these designs and compared them with CC analyses. Chow and Shao 7 developed an analysis method that is applicable to a design where two treatments are administered in sequences of three periods such that each subject completes at least two out of three periods. For their approach no assumptions on the distribution of the random effects in the common mixed-effects analysis model are required. It is not explicitly mentioned under which mechanism of missingness their analysis provides correct results. In fact, their estimator is a special case of the standard least-squares estimator (see Jones and Kenward, 8 p. 9), and hence the method described in Chow and Shao 7 would account for MCAR only.
None of the papers cited so far did consider the missing not at random (MNAR) situation which is not unlikely to happen. Richardson and Flack 6 examined to what extent the maximum-likelihood estimator fails under these circumstances. A recent paper by Basu and Santra 9 describes a model that includes the measurement and an outcome-dependent dropout process. However, their proposal looks like a Bayesian version of the Diggle and Kenward 10 approach. This paper attempted to estimate parameters from an informative missingness model to obtain evidence for MNAR. It also contains a simulation on the impact of MNAR on the analyses of a 4 × 4 cross-over trial with a Williams square design.
Since cross-over studies play a prominent role in drug development, for example, in BE trials, regulatory guidelines exist that propose analysis methods for these trials. The guideline made effective by CHMP11 in 2010 states the following: “The pharmacokinetic parameters under consideration should be analyzed using ANOVA … The statistical analysis should take into account sources of variation that can be reasonably assumed to have an effect on the response variable. The terms to be used in the ANOVA model are usually sequence, subject within sequence, period and formulation. Fixed effects, rather than random effects, should be used for all terms.”
The respective FDA guideline 12 recommends “For non-replicated crossover designs, this guidance recommends parametric (normal-theory) procedures to analyze log-transformed BA [bioavailability] measures. General linear model procedures available in PROC GLM in SAS or equivalent software are preferred, although linear mixed-effects model procedures can also be indicated for analysis of nonreplicated crossover studies. For example, for a conventional two-treatment, two-period, two-sequence (2 × 2) randomized crossover design, the statistical model typically includes factors accounting for the following sources of variation: sequence, subjects nested in sequences, period, and treatment … Linear mixed-effects model procedures, available in PROC MIXED in SAS or equivalent software, should be used for the analysis of replicated crossover studies for average BE.”
The CHMP guideline does not touch on the issue of missing observations at all. The FDA guideline does so only in the context of individual BE, but not for the concept of average BE which is applied most often in practice. To close this gap, this paper investigates the different approaches to cross-over trials in the context of incomplete data. The next section contains a review of the fundamental definitions of the missing data mechanisms and a statistical model for cross-over studies. In Section 3, we re-analyze the data from Chow and Shao 7 using fixed and mixed models and provide some simulations for the single sequence cross-over trial with incomplete data. Section 4 presents sensitivity analyses for the Chow and Shao 7 data.
2 Statistical background
2.1 Missingness mechanisms
In the following, we shortly review Rubin’s
13
taxonomy of missing data mechanisms. Let
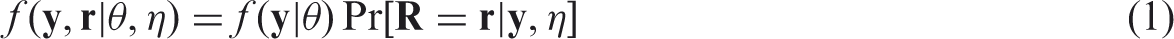
Alternatively, a pattern mixture model factorization can be obtained
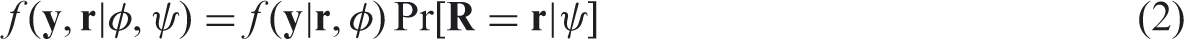
Note that (θ, η) has been replaced with
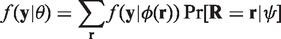
On the other hand, if the marginal distribution of
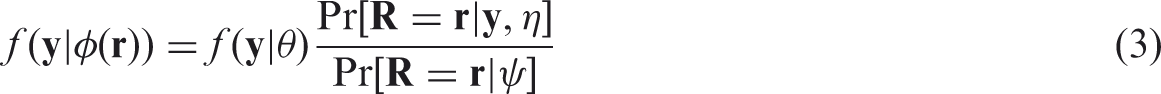
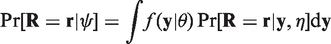
One obtains the distribution of the observed values by integrating out the missing observations. Doing this for the selection model (1) leads to
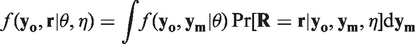
Under the MCAR mechanism the probability of an observation being missing is independent of the responses
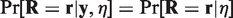
This implies
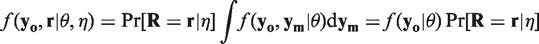
It follows from (3) that MCAR holds when all conditional distributions of the measurements given the missingness patterns are equal to the marginal distribution of the measurements and vice versa. In this case, selection and pattern mixture models are identical, that is,
For MAR the probability of missing depends only on observed data, that is

Here, we obtain
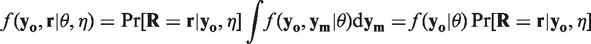
A straightforward consequence of MAR is that the conditional distribution of the missing observations given the observed measurements does not depend on the missingness pattern
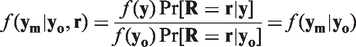
For both MCAR and MAR, inference can be based on the observed portion of the data, while the missingness mechanism can be ignored. Under this condition, likelihood-based analyses on the observed data are providing valid results when the caveats described in Kenward and Molenberghs 4 and Kenward 13 are considered. Particularly for MCAR, the analysis could be based on those units with complete information (CC analysis) since the missingness mechanism provides an independent random selection, although such an analysis would be inefficient.
When none of the above criteria holds then MNAR applies. In this case, the distribution of the missing data given the observed data and the missingness mechanism needs to be known for valid inferences, and both θ and η have to be estimable from the data
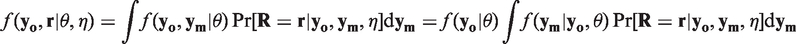
As in many other situations, assumptions have to be made on the distributions involved. However, in the MNAR situation these assumptions are principally not verifiable for all data, but only for the observed data. The best achievable is to analyze the data under a range of plausible alternative assumptions and to investigate the robustness of the results under these alternatives. An instructive example of a data-driven sensitivity analysis in the context of a selection model is Kenward. 14 Molenberghs et al. 15 considered a formal way of conducting sensitivity analyses by introducing influence analysis.
2.2 Cross-over studies
We consider general cross-over designs with p periods and t treatments. Let Yij denote the observation from subject i at period j. We model Yij as

It should be noted that in contrast to the recommendations in guidelines we do not account for a sequence effect for several reasons. First, for fixed-subject effects, a sequence effect would just confound the subject effect such that subject within sequence would have to be included into the model to render the sequence effect estimable. For random-subject effects, the inclusion of a sequence effect is discouraged, because this would be essentially self-contradictory: random effects are included to allow the incorporation of between-subject information on the mean effects, while fixed-sequence effects remove this information (see Kenward and Roger 16 ). Last and most importantly, subjects are randomly assigned to sequences and therefore by design the expected sequence effect is zero.
The random-subject effects are assumed to be identically and independently normally distributed with zero mean and covariance matrix Λ while the random errors are assumed to follow a normal distribution with zero mean and covariance matrix Δ not otherwise specified. Then the covariance matrix of the vector of observations from the i-th subject,

In the special case of the same random effect for all periods, that is, when subject is the only random effect in the model, uij = ui holds for all j. With


Parameter estimates are usually obtained from restricted maximum-likelihood estimation which allows to estimate the parameters of Σ in an unbiased way without having to estimate the fixed effects such as period or treatment effect first. Since the fixed effects are estimated given estimates of the variance parameters, their variability can be underestimated if the variability of the latter is not taken into consideration.
3 Mixed- versus fixed-effect models
3.1 Data from Chow and Shao
Here, we analyze the data presented in Chow and Shao, 7 which have been re-analyzed in Basu and Santra. 9 These data stem from a two-treatment, 3-period cross-over design where the treatment of period 2 in each of two sequences is repeated in period 3. Thirty-two patients were randomized into the first sequence and 34 into the second sequence. Missing data occurred only in period 3, namely, 8 for sequence 1 and 18 for sequence 2.
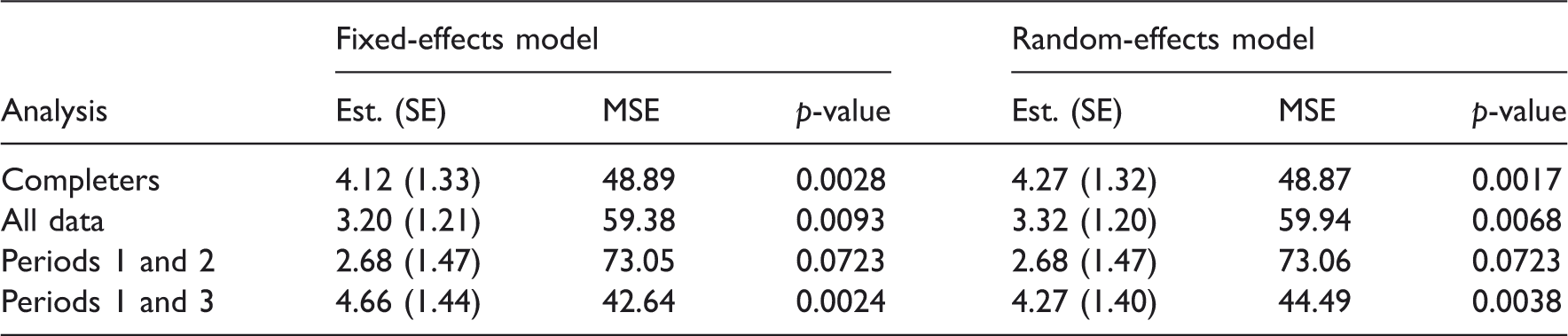
We run six different analyses for this data set based on model (4) with covariances (5) and (6) for the random effect and the random error, respectively. First, we consider completers only, that is, all patients with complete data from three periods, thereby reducing the total sample size from 68 to 42. In a second analysis, we consider only data from the first two periods to obtain 68 completers. Admittedly, nobody would add a third period to a cross-over trial and then ignore the data of that period entirely because some of them are missing, but this is done for illustrative purposes only. The third analysis considers all data obtained. We assume that there is no carry-over effect.
The results are obtained using a fixed-effects model (with treatment, period, and patient) and a mixed model (with sequence and treatment as fixed and patient as a random effect).
Estimates with standard errors, mean square errors (MSE), and p-values from different analyses of the Chow and Shao data.
3.2 Simulation of a single sequence cross-over trial
A design that is fairly often used to address the question of a potential DDI is the single sequence cross-over trial. Subjects receive drug A during period 1 and drug A and drug B during period 2 to assess the potential change in pharmacokinetics parameters of A in the presence of B. Note that carry-over effects are not an issue in these studies.
Results from 10,000 simulations of a single sequence trial (see text for details).
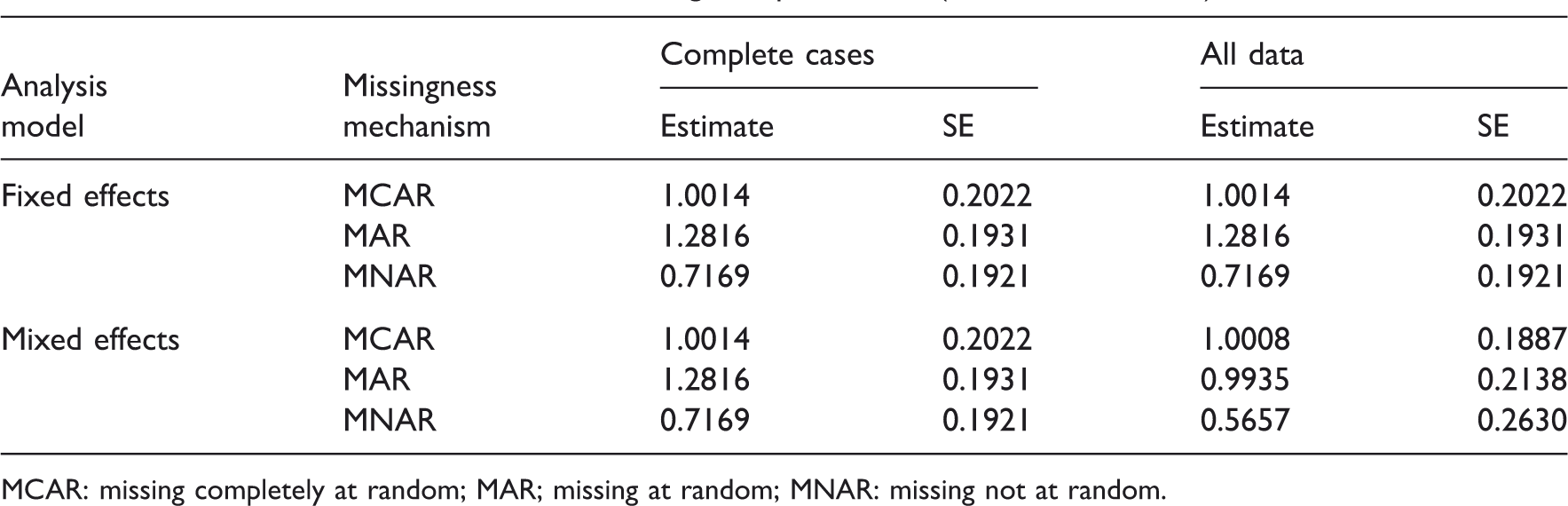
MCAR: missing completely at random; MAR; missing at random; MNAR: missing not at random.
As expected, all analyses fail to provide correct results under MNAR. The results for the fixed-effects model for CCs and all data are the same since the fixed-effects model cannot make use of the information from period 1 when data for the second are missing. The results are also the same as for the random-effects model for CCs since in this case the random effects are canceling out when the contrasts between treatments are formed. The mixed-effects analysis provides on average the correct estimates under MAR when all data are used and under MCAR when all data or just completers are considered. When one uses all data under MCAR, the standard error of the effect estimator is somewhat smaller than without using all the available data. The fixed-effects analysis provides sensible results only under MCAR and overestimates the effect under MAR.
These results call for a clarification of the statement that likelihood-based methods provide valid analyses under MAR. Both fixed-effects and mixed-effect models are likelihood based but only mixed-effects models provided the correct answer in the simulation above. In contrast to the mixed model, the fixed-effects model in the example above cannot use the information from the data of period 1 that predicted the missingness in period 2. Thus, a necessary condition (among others) for a likelihood-based method to provide correct results under MAR is that it utilizes all data points that predict missingness.
4 MNAR analyses based on the selection model
The previous sections have shown that there is a role for mixed-effect models in the analysis of cross-over trials, particularly in the presence of incomplete observations. In the latter case, mixed-effect models provide unbiased estimators of a treatment effect under MAR when the measurement model is correctly specified. However, though the MAR assumption may be reasonable in some cases it does not generally apply.
Under MNAR one needs to model the missingness process as well, not just the measurement process. It is tempting to fit such a model and let the data decide as to whether it fits better than a MAR model. However, such an approach ignores that a goodness-of-fit criterion can only assess the fit of the model to the observed data. Thus, evidence for or against MNAR can be provided solely within a particular predefined parametric family. In fact, every MNAR model can be doubled up with a uniquely defined MAR counterpart that depends on the same parameters and produces exactly the same fit as the original MNAR model. 19
4.1 The single sequence trial
We start with developing sensitivity analyses for the simplest case, that is, bivariate normal data (Yi1,Yi2) with mean 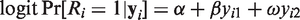
The parameter α reflects the extent of MCAR, β the extent of MAR, and ω the extent of MNAR in the missingness process. In particular, ω = 0 would imply that the missingness mechanism is ignorable. As said above, there is no intention to estimate ω from the data, but to vary it over a range of values to study the impact of MNAR on the estimation of the parameters of interest.
Let 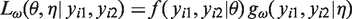
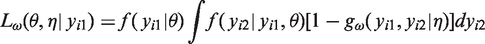
Note that 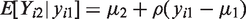
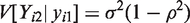
The likelihood is therefore given by
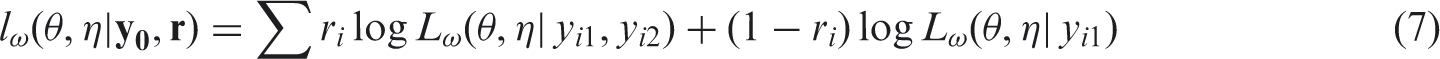
Sensitivity analyses can then be performed by obtaining parameter estimates of (θ,η) from maximizing (7) for different values of ω.
As an example we conducted such an analysis using the data from periods 2 and 3 of the Chow and Shao data set. Although the same treatments were applied during these periods within each sequence and therefore a zero difference should be expected, separate analyses were performed for the two sequences since the missingness mechanism could be treatment dependent. PROC NLMIXED of SAS
18
was used for the calculations since this software approximates the integrals in the missing data likelihood efficiently by adaptive Gauss–Hermite quadrature (see Liu and Pierce
20
). A graphical output of the analysis is depicted in Figures 1 and 2.
Estimates and 95% confidence intervals of the treatment differences Profile log-likelihood from periods 2 and 3 of the Chow–Shao data set for 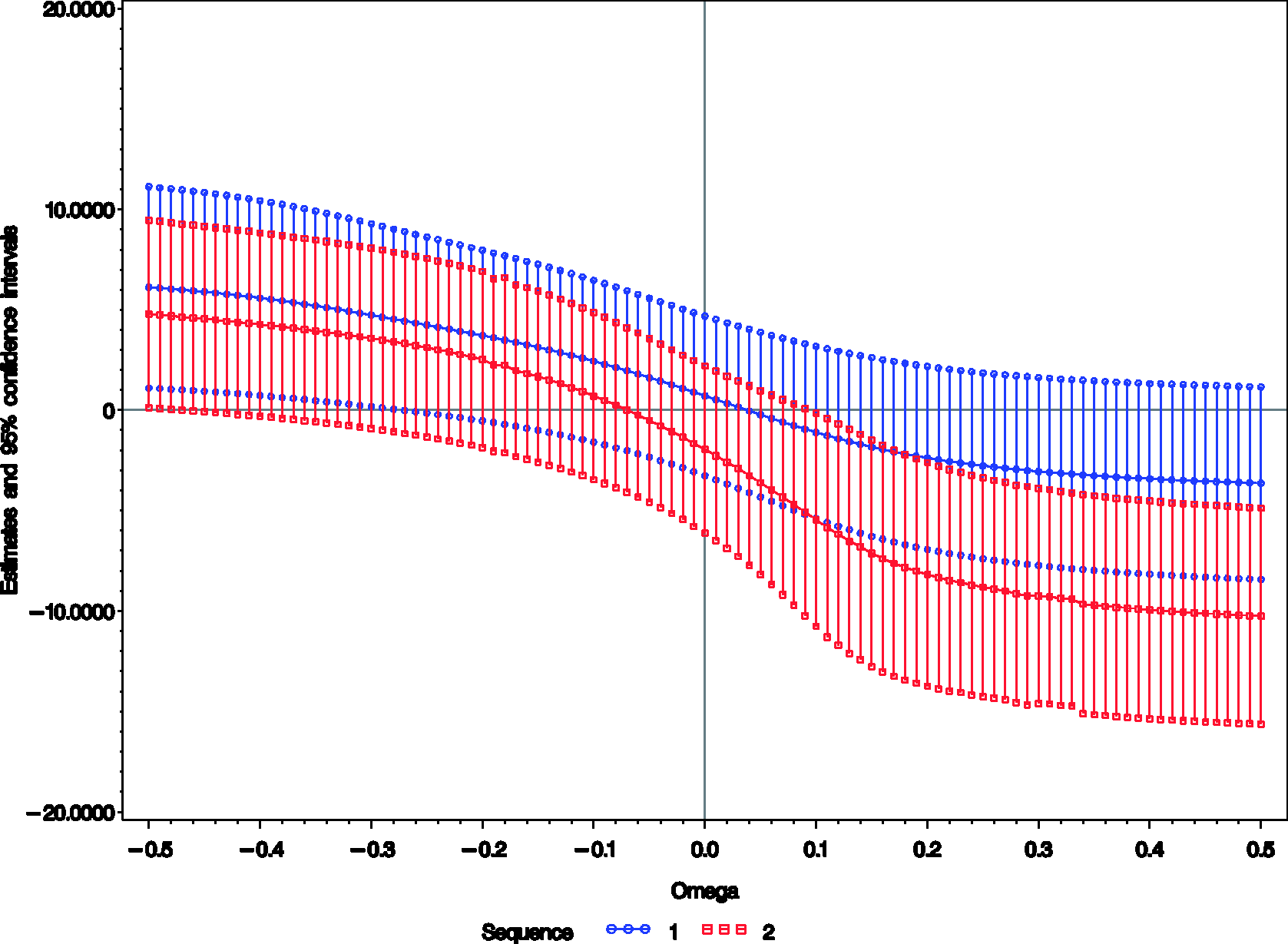
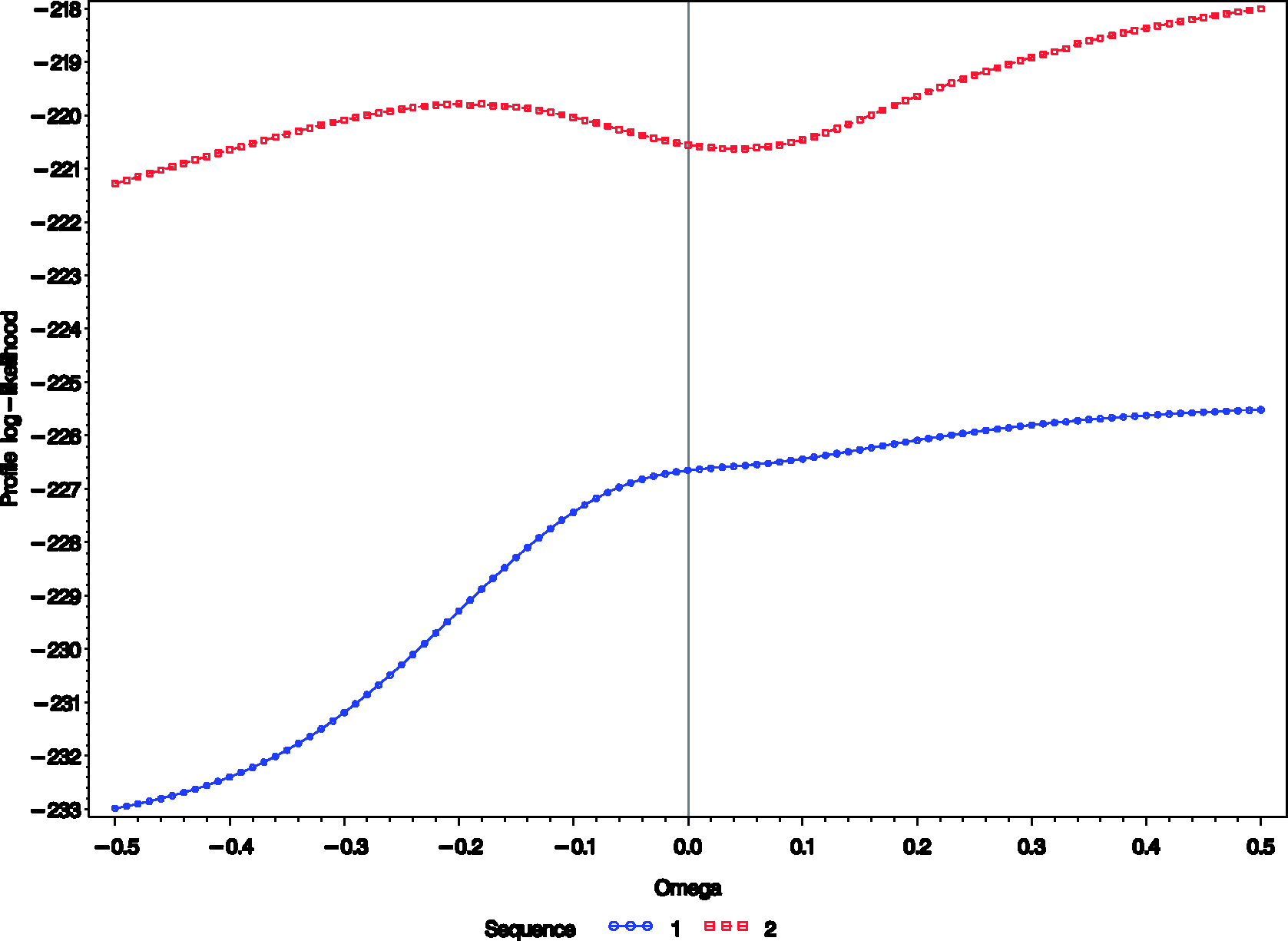
The treatment estimator for the second sequence seems to be more sensitive to MNAR than for the first, particularly for
4.2 The 2 × 2 cross-over trial
The considerations above can be easily generalized to cover the 2 × 2 cross-over design. Recalling the definitions in Section 2, we have
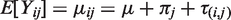
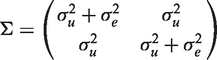
The equation for the mean implies that the distribution of 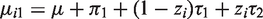
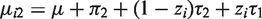
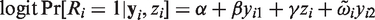
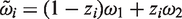
With 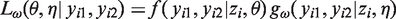
For an incomplete sequence we obtain
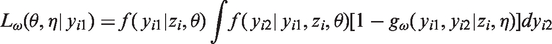
Note that 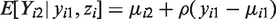
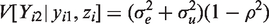
The full likelihood has then the same form as in equation (7).
The results of the sensitivity analysis for periods 1 and 3 data of the Chow and Shao data set are presented in Figures 3 and 4. For ω = 0, the estimate and the standard error for Contour plot of the estimates of the treatment differences Contour plot of the profile log-likelihood from periods 1 and 3 of the Chow–Shao data set (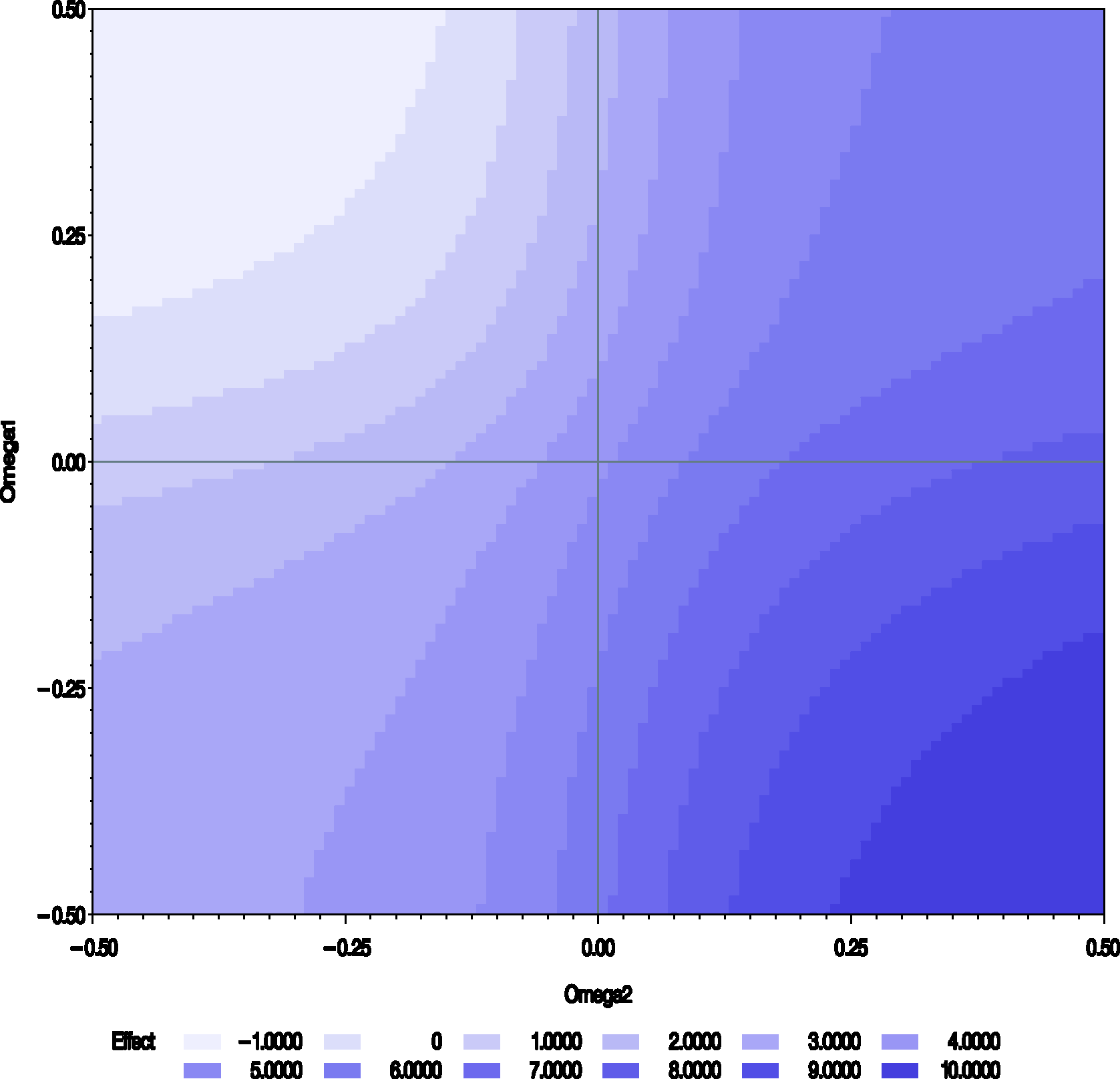
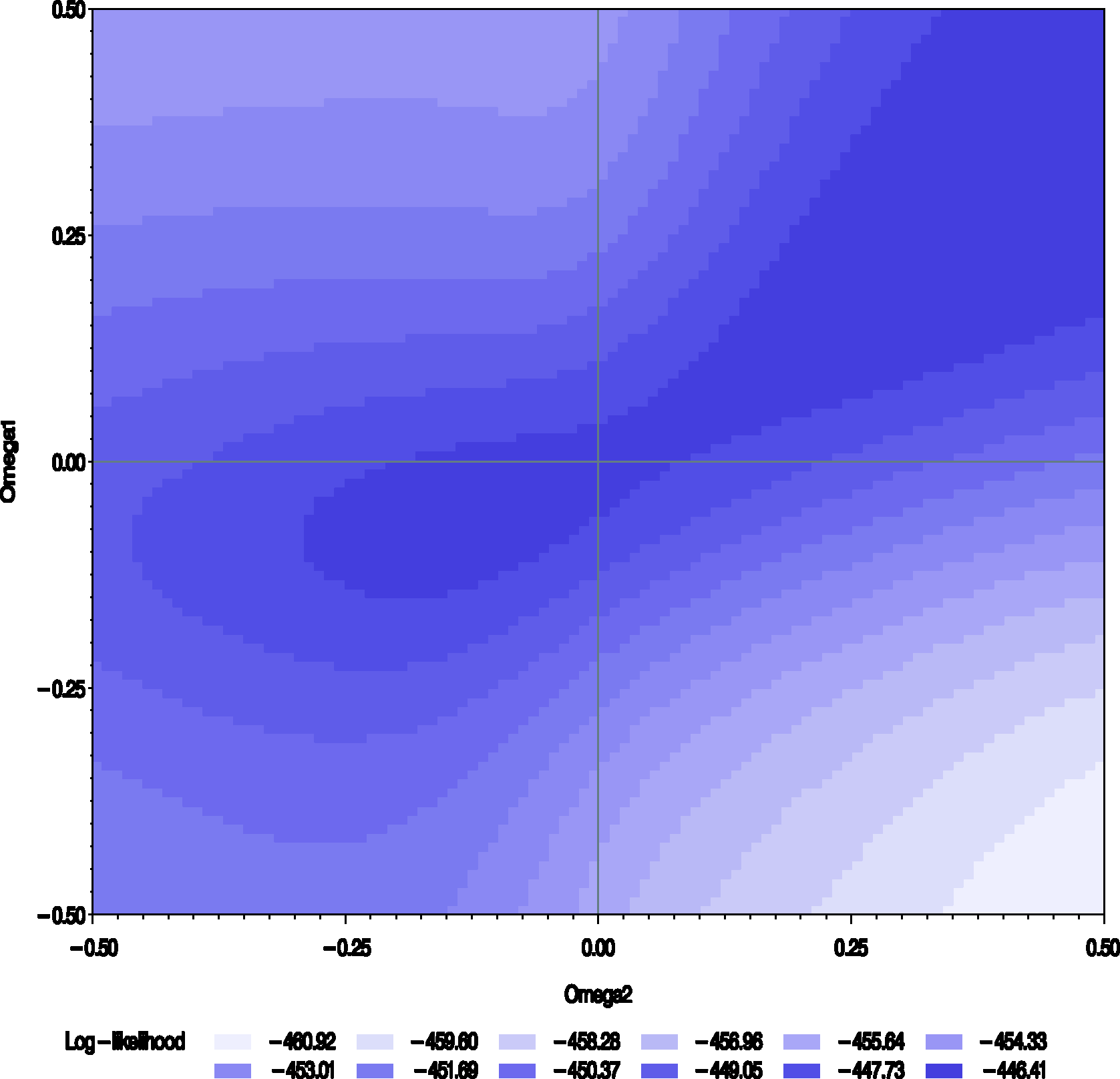
The profile likelihood has a maximum for values of ω around (0, 0) and along a stripe in the region where
5 Discussion
This paper has discussed aspects of the analysis of cross-over trials with incomplete data. In this context, we have argued that a mixed-effects model provides a valuable tool for the analyses of such trials. Consequently, recommendations and guidelines insisting on fixed-effect analyses may be unnecessarily rigid. When the model assumptions are appropriate, the mixed-model approach allows for a correct analysis under the MAR assumption by including all available measurements into the analysis, while the fixed-model analysis can only use within-subject information and provides a correct analysis only when MCAR holds.
Having said this it is fair to mention that one of the disadvantages of mixed-effects models is that the Wald statistics used to assess treatment effects are only approximately F-distributed and small sample corrections are required. The solution offered by Kenward and Roger 17 is implemented in PROC MIXED of SAS 18 and can be routinely used to relieve the issue.
In a well-designed cross-over trial without missing data, there is little information lost by discarding treatment information contained in the subject totals as done by using fixed-effects analyses. The introduction of random-subject effects into the model allows this information to be incorporated into the analysis when data are missing. In such an analysis, a weighted average is implicitly used that combines between- and within-subject effects. The weights are equal to the inverse of the variances of the two estimates (see Jones and Kenward, 8 Chapter 5).
Although MAR does not constitute a reasonable assumption in all cases it is the most general assumption under which a valid analysis is possible without considering the missingness mechanism explicitly. Such an analysis can serve as a starting point for further investigations of the dependency of the results under different assumptions on the missingness mechanism. We provided such sensitivity analyses using the selection model factorization. The principles applied can be taken forward to more complex cross-over settings as well. It would be interesting to see how a sensitivity analysis would look like in the pattern mixture framework, though as seen in Section 2, the assumptions made for one have direct implications on the other.
Footnotes
Acknowledgement
The author is grateful to Michael G Kenward, London School of Hygiene and Tropical Medicine, for offering his comments and sharing his insights during the preparation of this paper and to an anonymous reviewer for his diligent and constructive review of the submitted manuscript.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
None declared.