
Abstract
Background
Glia are the non-excitable cells of the brain, which, upon hyperstimulation, give rise to neuropsychiatric disorders. Recent evidence suggests that they are highly reactive cells, which makes them prone to environmental stimuli and early-life stressors. Upon excessive or chronic stimulation through exposure to stressors, these cells become responsible for causing neurological damage, which leads to neuropsychiatric disorders like schizophrenia, a global burden with no definite interventions.
Purpose
Perinatal stressors such as protein malnourishment, immunological disturbances, toxins, parental separation and abuse play a major role in negatively changing the cytoarchitecture and homeostasis of both neurons and glial cells (astrocytes, microglia and oligodendrocytes), finally degrading both cognitive and behavioural abilities. Effects of excessive glial activation and/or degeneration result in neuroinflammation, memory loss, anxiety, depression and hyperactivity-like symptoms in adult individuals, which contribute to the manifestation of schizophrenic pathology.
Conclusion
Therefore, it is believed that perinatal stressor-associated negative changes in glia can predispose an individual to develop this disorder later in life. This review summarises current evidence on how diverse early-life stressors influence glial cells, which could contribute to the neurobiological mechanism underlying schizophrenia.
Introduction
Recent studies have redefined the role of glial cells from being passive support elements to regulatory bodies of the nervous system. This category of cell includes astrocytes, oligodendrocytes and microglia, divided on the basis of origin, structure and function. Evidence suggests that astrocytes are the main supporting cells of neurons, which help in the regulation and formation of synapses, maintaining uptake and clearance of neurotransmitters, along with balancing the overall homeostasis of the brain. 1 Microglia, on the other hand, clear out unwanted moieties while protecting neurons and the brain from pathogens. Lastly, oligodendrocytes build and take care of the neuronal myelin sheath. Also, advances in organoid models, animal studies and transcriptomics have revealed diverse pathways through which glial cells regulate the neurons, helping them function properly. However, any stress-mediated alteration in these pathways causes irreversible changes in the neurons, which might be responsible for developing a very critical neuropsychiatric disorder named schizophrenia.1–3
The term schizophrenia, coined by Eugen Bleuler (1908), is characterised by both negative symptoms like depression, disorganised speech, low life expectancy, cell loss (neurons and glia) and abnormal motor coordination, as well as positive symptoms like hallucinations and delusions.3, 4 A schizophrenic patient often shows symptoms that are mostly residual with multiple episodes. 4 According to the WHO, schizophrenia is considered a global burden as it affected 24 million people worldwide in 2022. Its onset is seen during adolescence to early 20s in individuals who suffered severe mental distress during the developmental age. Schizophrenia is classified into five major types: paranoid, catatonic, hebephrenic, residual and undifferentiated schizophrenia.
The development of schizophrenia can be explained through three major theories: genetic, biopsychosocial and neurochemical theories. These theories contribute equally to the most acceptable hypothesis linked to the development of schizophrenia, that is, the neurodevelopmental model in combination with the dopamine hypothesis, suggesting a dysregulation of the dopamine system. However, schizophrenia can also be triggered by common factors such as perinatal infections, maternal malnourishment, physical and mental abuse, alcoholism, drug abuse and parental separation, the prevalence of which is increasing rapidly, and so is the risk for schizophrenia. Furthermore, according to the biopsychosocial theory, schizophrenia is a manifestation of interplay between social, biological and psychological factors. Thus, early-life stress in the form of abuse, neglect, infections, malnourishment and toxins can predispose an individual to develop schizophrenia.3, 4, 5
The above-mentioned stressors impact human life from a very early developmental age, also known as the critical window period. 3 Development of the nervous system in humans begins from the third week of gestation and is a protracted process that continues postnatally. 3 Formation, differentiation and migration of brain cells occur rapidly during specified perinatal periods of development via numerous well-orchestrated events, which are further modulated by intracellular, intercellular and environmental inputs. In rats, neurogenesis occurs between embryonic days (E) 10.5 to postnatal day (P) 15, with a peak at E14. Subsequent to the peak neurogenesis, there is a shift to astrogenesis. 1 Astrocytes are generated either from primary radial glia (RG), glia-restricted precursors (GRPs), secondary RGs of the subventricular zone (SVZ) or locally by proliferation of immature astrocytes.6, 7 Oligodendrocytes, on the other hand, are generated from oligodendrocytic precursor cells (OPCs), which are progeny of neural stem cells and generate oligodendrocytes during late embryonic stages and continue postnatally.8–10 Lastly, microglia originate from early embryonic erythromyeloid progenitors formed in the extraembryonic yolk sac (YS) around E7.5, and by E8.5, the cells become mobile and start migrating from the embryonic YS to the brain. Once the blood–brain barrier (BBB) is formed, the microglia become the permanent resident immune cells of the brain; they are shielded from peripheral toxicity to avoid unnecessary neuroinflammation.11–13 These crucial cellular events are negatively influenced by the early-life stressors, which disrupt healthy cellular development, leading to a compromised cytoarchitecture in the brain.
Astrocytes are the most predominant neuroglia, which regulate the brain homeostasis, alongside glycogen storage, glutamate uptake, regulation of ion concentration, vasomodulation, nervous system repair and formation of tripartite synapse. 9 Exposure to stress results in astrogliosis, leading to neuronal death and cognitive decline. Oligodendrocytes, on the other hand, produce neurotrophic factors and provide trophic support to the neurons by closely interacting with them. The saltatory transmission of nerve impulses in the mammalian nervous system depends solely on the myelin sheath, which is produced and maintained by oligodendrocytes through the process of myelination. 10 Myelin loss can be connected to oligodendrocytic degeneration following a pathogenic attack or stress exposure, and, in such conditions, the neuronal homeostasis is highly compromised,9, 10 leading to both cognitive and behavioural decline. On the other hand, microglia make up 5%–10% of the total brain glial population, which are in continuous surveillance for any abnormalities and cellular debris that need to be cleared through the mechanism of phagocytosis. While their main function is to clear debris, plaques and disintegrated cellular fragments, they also take part in spine engulfment and synaptic pruning. 12 The study of microglia in normal as well as in pathogenic conditions suggests that they can remain either in ramified (resting) or activated forms and respond drastically to any type of insult or adverse environmental conditions to actively regulate brain functioning. During healthy states, microglia are observed to communicate with other cell types via the exchange of cytokines, and upon activation, they are reported to upregulate the expression of significant marker proteins like Iba1 and MHC I/II, indicating their phagocytic activity. In inflammatory conditions, the activated microglia release a variety of inflammatory cytokines such as tumour necrosis factor alpha (TNF-α) (triggers neuronal apoptosis), interferon gamma (IFN-γ) (potent self-activator) and interleukin-8 (IL-8) (promotes growth and differentiation),12, 13 resulting in neuroinflammation and subsequent cognitive and behavioural decline.
Exposure to perinatal stressors disrupts the functioning of the glial cells and changes the homeostasis of the brain (Figure 1), leading to schizophrenia-like symptoms in the affected individuals. Perinatal malnourishment is the most common stressor that, in addition to a compromised nervous system, is also responsible for low body weight, muscle wasting and a poorly developed immune system in malnourished foetuses. In rats, maternal protein malnutrition leads to a delay in the formation and maturation of glial components, physical slowing, delayed reflex ontogeny, with permanent behavioural and cognitive impairments.14, 15 Additionally, viral and/or bacterial infections when encountered perinatally cause astrogliosis, microgliosis and neuroinflammation that accelerate the development of neuropsychological disorders. Furthermore, early life mental trauma and a toxic lifestyle such as exposure to alcohol and drug abuse superimpose an individual to schizophrenia. This review talks about the impact of the above-mentioned early-life stressors on brain cells and their correlation with the development of schizophrenia-like symptoms15, 16 (Figure 2).
Graphical Abstract.
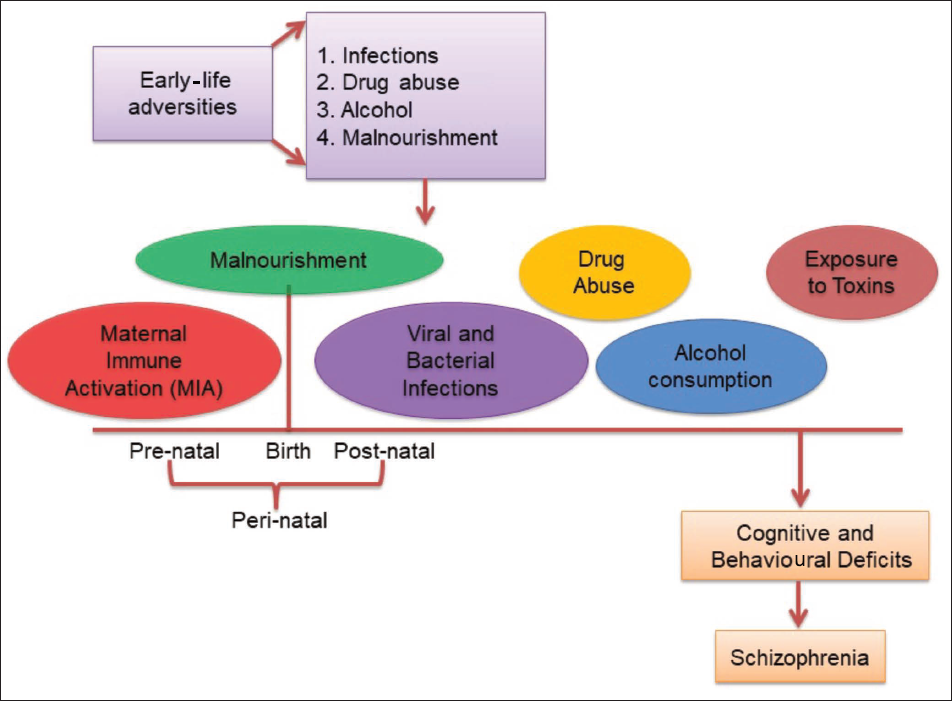
Impact of Early-life Stress on the Brain.
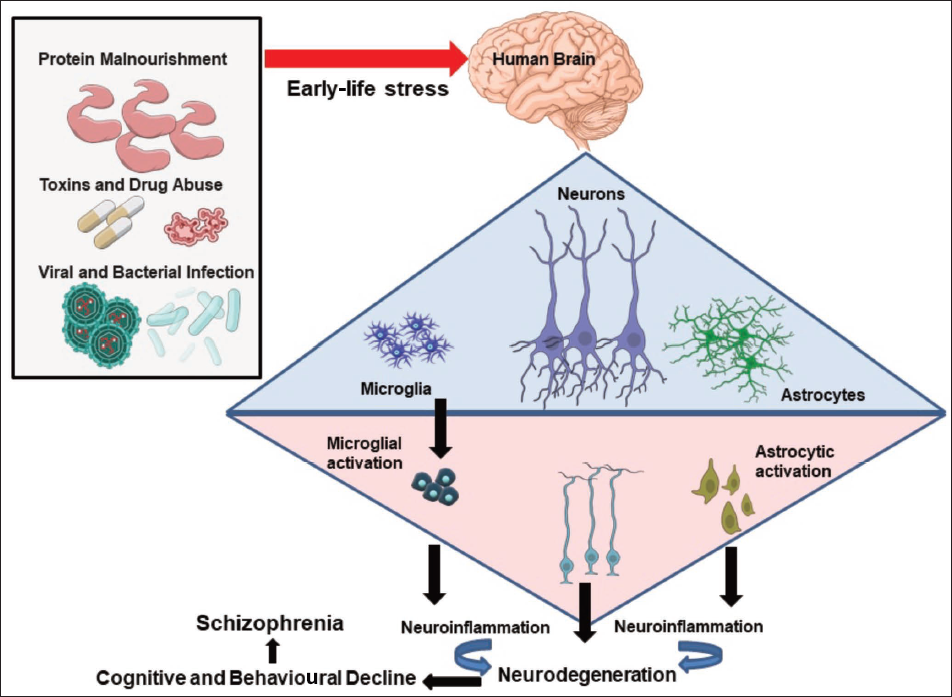
Perinatal Malnourishment, Glial Changes, Neurons and Schizophrenia
Malnourishment is the lack of essential nutrients, whereas undernourishment is the lack of sufficient calories in food. Both malnourishment and undernourishment give rise to a compromised physical, mental and immunological condition, which is further linked to schizophrenia-like symptoms in affected individuals. A study of the Chinese population who experienced famine (1959–1961) reported a correlation between lack of nutrition and schizophrenia, 16 whereas a magnetic resonance imaging (MRI) study of brains from schizophrenic patients exposed perinatally to the Dutch hunger demonstrated malnourishment-dependent changes in the brain. The patients were seen to have a compromised intracranial volume along with hyperintensity of the white matter. 17 Furthermore, studies16, 17 have reported the effect of prenatal malnourishment on the anatomy of the brain, such as shrinking of the corpus callosum, decreased diameter of myelin fibres and increased density of non-myelinated neurons, affecting neuronal conductivity. While protein malnourishment (PMN) majorly contributes towards a compromised physical and mental health, gestational PMN results in low birth rate and delay in cellular development, affecting both neuronal and glial proliferation along with behavioural impairments in later life. Evidence suggests that perinatal PMN predisposes affected individuals to develop neuropsychiatric and neurodegenerative disorders such as schizophrenia and depression during early adulthood, which mainly involves glial cells.18, 19
PMN affects microglia, making them unresponsive to external stimuli during development. Such microglial dormancy further affects neuronal plasticity because the formation and modulation of synapses require active involvement of microglia, failing which healthy synaptic pruning does not take place. 19 Also, perinatal PMN alters genes involved in inflammation and cell cycle in microglia, leading to poor cognitive outcomes. 20 A connection has also been established between nutrition and inflammation. Maternal PMN affects the immune function of the foetus and results in a diet-induced maternal immune activation (MIA). MIA transfers inflammatory cytokines to the foetus via the placenta, resulting in microglial priming through an increase in inflammatory cytokines like interleukin-1 (IL-1), interleukin-6 (IL-6), interleukin-10 (IL-10) and TNF-α.20–22 MIA, in turn, activates damage-associated molecular patterns (DAMPs) and toll-like receptors (TLRs), which is a major reason for microglial priming and neuroinflammation in malnourished individuals. 23 PMN-induced activated microglia, mainly in the grey matter, lead to the release of neurotoxic factors like free radicals, resulting in oxidative stress-related neuronal death and cognitive changes. Imbalance in antioxidant enzymes like superoxide dismutase, catalase and glutathione peroxidase, along with inflammatory cytokine-related neuroinflammation, results in causing symptoms of schizophrenia. Cognitive changes like learning and memory impairments are mostly related to synaptic loss, which is a consequence of excessive synaptic pruning by hyperactivated microglia. Thus, malnourishment-driven activation of microglia can be considered an important factor for schizophrenia.20, 21, 24
The impact of PMN is also seen in astrocytes, as it sharply reduces the A2B5+ glial-restricted progenitors (GRPs) in the ventricles and cortices of the developing brain, finally restricting the recruitment of GRPs to the cortex and subcortical structures. 25 PMN also restricts BLBP expression in the SVZ during early embryonic development (E14–E16), both in terms of expression and number of BLBP+ cells, hindering the formation of large-sized gliospheres and clusters, 26 which reflect the compromised astrogenesis in the developing brain. PMN during development was also reported to cause astrogliosis in the cortex, hippocampus and cerebellum, marked by an extracellular increase in S100β protein in adult rats.25, 26 Astrocytes also play a major role in maintaining the homeostasis of Ca2+ signalling, glutamate and dopamine pathways, which are considered impactful pathways that, upon interruption, lead to schizophrenia-like symptoms. PMN results in both astrocytic death as well as abnormal astrogliosis, which subsequently cause synaptic loss, glutamate toxicity and neuronal death. Such events lead to a compromised cognitive ability as well as behavioural deficits like anxiety and hyperactivity. Additionally, in tripartite synapses, PMN causes a decrease in serine, glycine and glucose metabolism, which causes an imbalance in excitatory and inhibitory neuronal signalling, resulting in schizophrenia-centred behavioural deficits.20, 21, 26
Furthermore, the impact of PMN on glial cells can be understood through studies which show that intra-generational PMN delays cellular differentiation, maturation and activation, further inducing poor habituation-with-time in novel environment exploration, low anxiety and hyperactive profiles and poor forelimb neuromuscular strength in F1 generation of the protein malnourished rat mothers, which corresponds well with the hallmarks of neuropsychiatric diseases like schizophrenia.26, 27 Studies which specifically dealt with PMN also stated that it alters both the quantity and quality of myelination in terms of decreased myelin binding protein (MBP) expression, affecting myelination and neuronal connectivity. 27 Such changes in the myelination status are mostly due to decrease in oligodendrocytic precursors with concomitantly downregulated myelin proteins like MAG and MOG in perinatally protein-malnourished F1 rats. 28 Also, maternal protein deprivation significantly lowered platelet-derived growth factor receptor-α (PDGFRα) oligodendrocyte precursor cells (OPCs) in the rat model and negatively influenced expression of myelin protein genes. This caused a decline in myelinating oligodendrocyte population, hypomyelination and misaligned myelinated fibres, leading to long-term changes in the corpus callosum and demyelinating lesions in the brains of F1 pups.27, 28 Such myelination deficits have also been observed to result in behavioural deficits seen in schizophrenic brain. A mutation in oligodendrocytic genes responsible for myelination results in a decrease of myelin proteins, causing compromised myelination of neurons. PMN also results in oxidative stress-induced myelin damage, which is responsible for behavioural deficits common in schizophrenic patients.28, 29
The above-mentioned changes in glial cells following PMN finally affect the cytoarchitecture of neurons, further compromising the neuroanatomy of the brain. Additionally, concentrating on the direct impact of PMN on neurons, it increases oxidative stress by increasing oxidants like lipid peroxidase, resulting in neurodegeneration. 20 Such oxidative stress is common in malnourished rats showing schizophrenia-like pathophysiology. Another mechanism of action of prenatal nutritional deficiency is reported to be downregulation of underlying genes like Dlgap3, Grm5, Furin and Ubqln1. The mentioned genes regulate synapse formation and cognitive functioning, and their dysregulation results in schizophrenia-like symptoms in affected individuals. Malnutrition is also observed to disrupt neurogenesis. Through immunohistochemical analysis, it was found that PMN decreases neuronal progenitor cells (NPCs) in protein-deficient rats, suggesting an impaired process of neurogenesis.24, 30 These changes in neurogenesis, neuronal cytoarchitecture and neuronal connectivity eventually cause changes in cognitive and behavioural abnormalities, leading to schizophrenic conditions. 20
Furthermore, intrauterine growth restriction (IUGR) and micronutrient deficiency also result in high morbidity in later life with drastic changes in learning and memory, social behaviour, poor motor coordination, decreased IQ scores and impaired school performance. Bioactive nutrients like folate and methionine epigenetically modify the DNA through methylation and change the pattern of gene expression in the dopaminergic, neurogenesis and BDNF pathways. These changes further alter the cognitive and behavioural abilities.20, 21, 29, 30 Studies have reported that deficiency of calories, folate, vitamin D, essential fatty acids, retinoids and iron occurs in offspring that suffer maternal malnourishment. Such conditions alter the expression of genes specific to Alzheimer’s disease and schizophrenia, further predisposing an individual to develop psychotic disorders. 31
Glial and Neuronal Changes Following Viral and Bacterial Attack in Correlation to Schizophrenia
Glial changes are common following viral and bacterial infections. Such infections are prevalent in places with poor hygiene and healthcare systems. Many viruses (Herpes simplex, Herpes zoster, cytomegalovirus and West Nile virus) and bacteria (Neisseria meningitidis and Streptococcus pneumoniae) attack a wide range of populations at an early age, giving rise to inflammatory conditions in the brain causing meningitis and encephalitis. 32 Lifelong impairments in the neuroimmune and neuroendocrine responses in rodents have been related to early-life infections. Perinatal induction of influenza in mice has been used as a model to study schizophrenia-like situation. 33
Glial and Neuronal Changes Following Viral Stress in Relation to Schizophrenia
Viral infections are linked to encephalitis, but recent studies have reported a wider aspect of viral infections and neurological disorders. Synthetic double-stranded RNA and a viral mimetic, that is, polyinosinic:polycytidylic acid (poly I:C), exposure during the gestation period induces expression of pro-inflammatory cytokines such as interleukin-1β (IL-1β), IL-6 and TNF-α. Poly I:C has been established as a neurodevelopmental model of schizophrenia,32, 33 and such inflammatory responses have direct relevance for decreased motor activity and motor coordination. 33
Early-life viral infection impacts the glial cells majorly and abnormally accelerates microglial proliferation and activation, disrupting healthy microglial activities like synaptic pruning and phagocytosis, which leads to an imbalance in the glial and neuronal homeostasis. Microglia, being the immune-competent cells of the immature brain, are sensitive to infections 34 and actively cause neuroinflammation by hypersecretion of pro-inflammatory cytokines. Studies have reported that poly I:C is responsible for inducing MIA in mice, which increases the expression of both pro- and anti-inflammatory cytokines, chemokines, as well as colony-stimulating units in the brain of both mother and offspring. Such hypersecretion of inflammatory moieties occurs as a result of microglial activation, which is higher in the case of neonates when compared to adults, suggesting that early-life viral infections potentially cause long-term behavioural and cognitive impairment mimicking symptoms similar to schizophrenia. Additionally, early-life viral infections are also reported to trigger cellular apoptosis in the brain, which in turn hyperstimulates microglia, leading to synergistic damage in the central nervous system (CNS).34–36 Once activated, microglia are localised around the site of inflammation, releasing cytokines, prostaglandins, matrix metalloproteases and NO, further causing neurotoxicity and contributing to neurodegeneration. Thus, microglia can be considered a potent contributor to both positive and negative schizophrenic symptoms upon activation by viral attack.
Again, viral infections also impact astrocytes, which are in close contact with neurons, blood capillaries, ventricle cavities and pia mater. Some viruses, being neurotrophic, can invade the nervous tissue through retrograde transport, damage the endothelial cells by BBB breaching and trigger leucocytes to infiltrate into the brain through the damaged blood–cerebrospinal fluid (CSF) barrier. Astrocytes are the major source of IFN in the brain and, on activation, produce cytokines and chemokines that restrict viral growth and protect brain cells.
37
Studies using early-life viral-infected rat models created by poly I:C administration to pups reported astrocytic changes in different brain regions like the cortex and hippocampus. Astrocytes upon early-life threat undergo astrogliosis, marked by the upregulation of specific marker proteins like GFAP, S100β and vimentin.
38
Poly I:C is recognised by TLR3 receptors via the nuclear factor kappa B (NF-κB) pathway, and oversensitisation of TLR3 receptors in neonates is found to increase vulnerability to hypoxia-ischemia via TRIF (toll/IL-1R domain-containing adaptor molecule-1) dependent inflammatory response.32, 37, 38 MIA with poly I:C is also found responsible for causing sickness behaviour in mothers as well as astrogliosis and memory impairment in offspring. Astrocytes on activation via poly I:C are reported to secrete mediators like IL-6, CCL2 and CXCL10, which trigger immune inflammation leading to cognitive and behavioural impairment, common in schizophrenic patients.32, 34
Virus can directly attack and damage the myelin sheath, but some viruses, like Theiler’s virus, attack the neurons initially and then reach the cytoplasmic channel of the myelin sheath, leading to a persistent viral infection. 39 Viral infection might cause demyelination, giving rise to multiple sclerosis. However, myelination continues postnatally, and hence the perinatal period is vulnerable to demyelinating disorders, which in turn affect the motor abilities of the affected individuals. Demyelination occurs either as an immune attack on the myelin sheath or due to oligodendrocyte death. Common viruses like influenza easily target neonates and affect myelination by altering MBP. 40 In the case of Zika virus infection, it was found that myelinating oligodendrocytes are more easily attacked by this virus, which makes the patient susceptible to demyelinating disorders. 40 Poly I:C-induced microglial activation also promotes oligodendrocytic death through TNF-α/TNFR1-dependent pathways, further affecting the myelin sheath. Furthermore, MIA in mice using poly I:C was also reported to delay myelination in pups with significant downregulation of MBP protein in the affected offspring. 41
Lastly, all these glial changes following viral infection lead to neuronal deterioration. Both neuroinvasive and neurotrophic viruses, such as herpes simplex virus 1 and 2, cytomegalovirus, Epstein–Barr virus and coronavirus, can attack the neurons by binding to different neuronal receptors and contribute to neuroinflammation and schizophrenia-like conditions. 42 Again, a combined study has reported that both MIA induced through poly I:C exposure and herpes simplex virus 1-mediated infection in neuronal precursor cells have shown a downregulation of schizophrenia-related genes like PCDHA2, PCDHA3 and PCDHA5. These genes are associated with neuronal cell adhesion, and their downregulation could be the reason for schizophrenia-related phenotypes.43, 44 Additionally, during COVID-19 and related pandemic, a link between viral infection and psychosis was observed, and there are reports which explain the direct and indirect mechanisms through which viruses impact the neuronal cytoarchitecture.44, 45 Thus, neuronal impairment following viral infection could be one of the reasons for schizophrenia-like symptoms in affected individuals.
Glial and Neuronal Changes Following Bacterial Stress in Relation to Schizophrenia
Bacterial infections are very common during early life and are reported to reach the nervous system in severe cases, causing bacterial meningitis. The mechanism by which a bacterial infection alters the nervous system includes the involvement of glial cells. The mode of action of bacteria can be diverse, but most often, bacteria circulating in the bloodstream enter the brain either directly (transcellularly via endothelial cells or paracellularly via cell junctions) or indirectly (Trojan horse method via infected phagocytic cells). 46 On pathogenic attack, glial cells, mainly microglia, respond as an element of the defence mechanism of the nervous system and limit the damage. But in case of chronic early-life infections, the microglia become oversensitised, leading to hyperproduction and release of pro-inflammatory cytokines, and such conditions give rise to inflammation of the nervous tissues, concomitantly causing glial death and neurodegeneration. Following lipopolysaccharide (LPS; bacterial endotoxin) administration at gestation day 3 and 5, Sarkar and co-workers observed activated microglia with increased density and proliferation. In addition, such microglia also presented macrophage-like properties with inflammatory responses and antigen presentation, a threat to the developing brain. LPS has been reported to increase tyrosine kinase and protein kinase C activity, increasing reactive oxygen species (ROS) signalling and releasing inflammatory molecules via microglia.20, 46–48 Also, on LPS induction, microglia go through morphological changes and transform from a ramified or resting state to an amoeboid or activated state, and such phenotypic activated state is accompanied by an increase in levels of marker proteins like Iba1, OX6 and OX42. On stimulation during early age, microglia conduct their phagocytic activities and lead to intracellular bacterial killing along with glial death, leading to memory impairment in adulthood.49–52 Such early-life glial activation is also found to be responsible for the development of anxiety and hyperactivity, which finally results in disorders like schizophrenia.
Bacterial invasion also attacks the astrocytes by disrupting the BBB and triggers the immune response. Astrocytes perceive bacterial antigens via microbial pattern recognition receptors, TLRs (TLR2, 4, 5 and 9) and nucleotide-binding domain leucine-rich repeat region. 53 After recognition, they react as part of the innate immune response and recruit peripheral immune cells to trigger adaptive immunity. Astrocytes are believed to cluster around the injured tissue to restrict the local damage, giving a scar-like appearance. LPS, as mentioned above, is found to increase the GFAP expression in an astrocytic cell culture, thereby indicating astrogliosis. The mechanism behind such LPS-mediated activation of astrocytes is the disrupted Ca2+ level and signalling pathway.54, 55 On activation, the astrocytes are reported to increase production of cytokines, NO and cyclooxygenase enzyme (COX2), increasing the enzymatic activity and the expression of LPS receptor (CD14) on the surface of the activated astrocytes. Studies have also reported an increase in the proliferation of astrocytes typified as reactive astrogliosis on early-life bacterial stimulation. Such astrogliosis is marked by an increase in expression of astrocytic marker proteins GFAP and S100β. LPS also acts on astrocytes by increasing the release of NO and NO synthase activity, which in turn contributes to the degeneration of oligodendrocytes and neurons, further causing myelin sheath and synaptic damage, leading to a neurodegenerative condition. 56
Oligodendrocytes are also impacted by bacterial attack. Bacterial infection affects the normal myelination procedures by interfering with both oligodendrocytic proliferation and maturation 57 and is related to various demyelination diseases including multiple sclerosis. Studies have reported that neonatal LPS exposure downregulates myelin proteins like MAG, MOG and MBP, leading to loss of the myelin sheath. 58 Studies attempting to explain the early-life infection and later-life impact on myelination and behaviour in rodents have also noted that demyelination during early life, that is, 3–6 months of age, leads to white matter damage in the corpus callosum and cortex at later age (12–24 months), with downregulation of MOG, leading to the loss of myelin compaction. Such demyelinating conditions are related to many neurological disorders including schizophrenia. Findings also suggest that bacterial insult during developmental period results in increased apoptosis of oligodendrocytes, delaying the process of myelination. Such oligodendrocytic death occurs due to hypersecretion of NO and inflammatory cytokines by activated microglia and astrocytes, which in turn modulate the expression of myelin-related genes like MBP and MOG, leading to demyelination and oligodendrocytic death, affecting motor abilities in affected individuals. Such abnormalities are related to the development of later-life schizophrenic conditions.56, 58, 59
Lastly, all the glial damage following bacterial infections impairs the neurons, and studies have reported that LPS exposure during pregnancy in rats resulted in offspring with hyperactivity and prepulse inhibition impairment. These behavioural impairments were followed by recognition memory deficits and loss of myelin sheath, connecting bacterial infection to schizophrenia. Schizophrenic patients produce low levels of glutamic acid decarboxylase (GAD67) and its respective GABA neurons. Prenatal LPS exposures in rats are seen to lower the GAD67+ cells in the hippocampus. Additionally, MIA following LPS exposure is reported to alter the dopamine system, contributing to schizophrenia-like conditions. MIA interferes with the development of the foetal brain through inflammatory cytokines, antibodies and immune cells. MIA following LPS exposure disrupts neuron–microglial crosstalk via alternation of CD40, IL-1β, TNF-α, Arg1, Iba1 and others, further getting involved in the aetiology of schizophrenia.60, 61 Thus, the relation between bacterial infection and glia-mediated neuronal changes cannot be neglected and could be one of the major reasons for the development of schizophrenia.
Unhealthy Lifestyle, Toxins and Development of Schizophrenia
Alcohol and Glial Cells of the Central Nervous System
Apart from poor diet, unhealthy lifestyle like consumption of alcohol, especially during pregnancy, can have negative effects on both glial and neonatal health, causing foetal alcohol syndrome. Maternal alcohol consumption affects gliogenesis and interrupts glial–neuronal interaction in the foetus. Alcohol exposure during the development of the immune system can also lead to microglial activation, which can cause foetal alcohol spectrum disorders and long-term cognitive and behavioural impairment in affected individuals. 62 Foetal alcohol exposure is also reported to cause neurotoxicity, thereby activating microglia. Such activated microglia gather around dead cells and engulf the debris. However, on chronic alcohol exposure, the death rate of neurons was found to exceed the clearance capacity of microglia, increasing the burden of inflammatory cytokines in the developing brain. 63 Moreover, it was seen that alcohol intake during adolescence leads to partial activation of microglia in the hippocampus, suggesting long-term changes in the brain. 64 Alcohol exposure during development or adolescence also causes oxidative stress, resulting in neuronal apoptosis and vigorous microglial activation with long-term cognitive and behavioural impairment. 65 Additionally, exposure to abused drugs also imposes a long-term impact on the microglial profile. For example, morphine exposure during adolescence is reported to increase TLR4 receptors in microglia and cause microglial activation. Cocaine is also observed to activate microglia and elevate TNF-α levels, which further results in decreased synaptic strength and behavioural sensitisation. Apart from alcohol and drug abuse, early-life adversities in the form of parental separation are also seen to sensitise microglia, leading to their activation. Improper parental care also results in improper development of the innate immune system, changing microglia activities and resulting in abnormal maturation of neuronal processes, causing the risk for schizophrenia.66–68
Furthermore, ethanol is also known to delay astrocytic differentiation, migration and protein trafficking. A significant decrease in GFAP expression was seen in ethanol-treated individuals, further suggesting its impact on gliogenesis and the risk of developing schizophrenia in affected individuals. 69 Alcohol also hampers the development of radial glial cells and reduces the secretion of growth factors by astrocytes, causing improper neuronal development and oxidative stress, further leading to degeneration of neurons in affected individuals. 70 Although limited studies are available that correlate alcohol consumption and astrocytic activity, it cannot be overseen that overall astrocytic activity and morphology are affected following exposure to alcohol at a very early age. Such astrocytic changes are directly related to neuronal and synaptic damage, which are mainly responsible for memory loss and learning deficits.70, 71
Oligodendrocyte functioning is important for the healthy transmission of the electrical potential among neurons. Any change in this glial subtype results in behavioural deficits, which contribute to schizophrenia-like symptoms in affected individuals. The in utero environment is another important factor for proper oligodendrocytic development. When a non-human primate foetal brain was exposed to alcohol in the third trimester, apoptosis of cells in the white matter was observed, and these cells were mostly oligodendrocytes.72, 73 Postnatal alcohol exposure in rats also delayed oligodendrocytic maturation, causing negative effects on later-life motor abilities. Ethanol is also reported to cause white matter degeneration in rats, impairing oligodendrocytic functioning and myelination. Such abnormalities during developing age can thus contribute to the development of foetal alcohol spectrum disorders. 74 Schizophrenia can also be caused by early-life drug abuse, and in such cases, oligodendrocytes are highly affected. Among drugs, cocaine and opioids are reported to bring out changes in the expression of myelin-related genes (PLP1, MBP and MOBP), alongside decreasing the overall population of oligodendrocytes. This results in white matter pathology, altering myelination, cognitive and behavioural abilities in abused individuals.73, 74
Lastly, schizophrenia-like symptoms are also linked to defects in the dopaminergic system, and alcohol abuse is reported to stimulate dopamine release, associating it with the positive symptoms of schizophrenia. Postmortem studies of both alcoholic and schizophrenic brains have identified a decrease in volume of both pons and thalamus. Whereas alcohol misuse is reported to decrease neuronal spine density and pyramidal neuronal soma volume, which further decreases the efficacy of neurons, leading an individual to show symptoms similar to schizophrenia patients. Furthermore, alcohol dependency also interferes with the emotional centre of the brain, that is, amygdala by increasing GABAergic transmission and inhibiting the glutamatergic transmission via NMDA and AMPA receptors, resulting in behavioural impairments.66, 75–78 Thus, all these mentioned reports point towards a connection between alcoholism and neurological disorders like schizophrenia.
Toxins and Glial Cells
Our early life is in close contact with unavoidable artificial agents, heavy metals, chemicals and toxins. However, such harmful toxins can have a negative impact on the brain cells, mainly glia. Among the glial subtypes, microglia actively respond to environmental toxins and in turn are responsible for the development of neurotoxicity in the CNS. Toxins, both in the blood as well as in the nervous system, activate microglia via pattern recognition receptors, which generate ROS in the nervous system. These ROS further cause self-activation of microglia, which release inflammatory cytokines, creating an imbalance between pro- and anti-inflammatory cytokines and leading to neurodegeneration.79, 80 Also, such ROS generated during chronic activation of microglia was reported to cause loss of dopaminergic neurons in the substantia nigra and developing Parkinson’s disease (PD) in affected individuals. Additionally, MIA via environmental toxins such as lead, mercury and arsenic can also accelerate the development of autism spectrum disorders (ASD) in affected neonates via vigorous activation of microglia and chronic neuroinflammation. Pesticide exposure (paraquat and dieldrin) is also reported to contribute to neurodegeneration and PD-type pathology by activating microglia through the NOX2 pathway (NADPH oxidase 2), which further leads to the generation of ROS.81–84 While specifically talking about schizophrenia-like condition, early life exposure to heavy metals results in hyperactivation of the innate immunity by causing oxidative damage to the cells. Such a response leads to hyperactivation of the microglia, resulting in the accumulation of pro-inflammatory cytokines. These cytokines cause neuroinflammation and related cognitive and behavioural impairment, which is also seen in schizophrenia.
Astrocytes are also commonly affected by exposure to harmful environmental toxins, giving rise to nervous system-related disorders. Millions of people worldwide come in contact with arsenic, a carcinogen which potentially causes neurotoxicity. Arsenic is reported to cross the placenta and affect the foetus in pregnant mothers. It can also cross the BBB and reach the brain cells in a dose-dependent manner, causing apoptosis of astrocytes by disrupting the GFAP cytoskeletal proteins of the cell. Lead (Pb) is another such heavy metal that contributes to neurotoxicity by blocking NMDA receptors and inhibiting glutamatergic transmissions, which then further leads to accumulation of excess glutamate, leading to glutamate toxicity. 85 Aluminium toxicity also causes astrocytic death by apoptosis and toxicity attack, further leading to loss of neurons. Many studies have also linked PD and amyotrophic lateral sclerosis (ALS) with environmental conditions. While an enriched environment can protect neurons from degeneration, early-life exposure to different toxins (deltamethrin, cobalt and food contaminants like T-2 toxin) activates caspase pathways and causes astrocytic death, further developing neurotoxicity and neuronal degeneration.86, 87
Environmental toxins can also cause demyelinating diseases. Early life is prone to toxins, making oligodendrocytes an easy target, which further compromises the myelination process. Some biological toxins like diphtheria toxin lead to diphtheric neuropathy, characterised by vacuole formation and myelin damage. 88 Cuprizone, a copper-chelating mitochondrial toxin, is responsible for triggering apoptosis of oligodendrocytes in the corpus callosum by altering the activity of superoxide and complex IV of mitochondria and causing demyelination-related disorders.87, 88 Pesticides used in agriculture can also have a negative effect on the myelin sheath. Organophosphates are widely used as pesticides and are considered safe for humans. However, studies have reported that chlorpyrifos is a new organophosphate which is no longer considered safe. Juveniles are considered to be more prone to this chemical when compared to adult individuals. It is a potent neurotoxin and is also toxic to oligodendrocyte progenitor cells. Chlorpyrifos is also reported to induce oxidative stress and activate caspase pathways, leading to cellular apoptosis. 89 Such early-life impact on myelination also causes cognitive and behavioural deficits, leading to a risk for neurodegenerative disorders.
Environmental risk factors like exposure to toxins, mainly during the critical period of development, influence the cytoarchitecture of neurons. Early-life exposure to lead can be considered responsible for triggering psychotic disorders like schizophrenia because lead is found to alter the NMDA receptors and cause their hypofunctioning. Lead neurotoxicity is also reported, stating that lead exposure alters the cognitive and behavioural impairment in exposed individuals mainly by changing the dynamics of calcium, neurotransmitters and neuronal homeostasis. Furthermore, neuronal damage is also seen in the case of mercury toxicity via depletion of glutathione in the CNS.90–92 Studies have also reported that heavy metals such as mercury, lead and aluminium can accumulate in the locus coeruleus neurons upon exposure and cause weakening of the BBB. In fact, pesticides are also found to directly harm neurons and cause mental disorders. Fipronil, a phenylpyrazole pesticide, is reported to inhibit GABA in rat dopaminergic neurons, altering the dopamine network and resulting in neuropsychiatric disorders. 90
Impact of Cellular Changes on Cognitive and Behavioural Abilities with Respect to Schizophrenia
The overall changes in the brain cells due to early-life stress exposure disrupt the healthy neuron–glia cross-talk and trigger the development of several neurological disorders. Such neurological disorders are marked by cognitive and behavioural deficits, which are studied as phenotypes for the same. The negative aspect of schizophrenia is mainly marked by depression, triggered by both deficiencies (low levels of omega-3, vitamins and minerals) as well as cellular inflammation.91, 92 Cellular inflammation caused by both viral and bacterial infections increases the level of pro-inflammatory cytokines in the brain, among which IL-6 is reported to be responsible for initiating symptoms of schizophrenia. Also, excessive activity in the hypothalamic–pituitary–adrenal (HPA) axis is also reported during chronic stress, which in the long term results in negative symptoms of schizophrenia.92, 93 Anxiety, on the other hand, is another psychological marker of schizophrenia that occurs due to an imbalance in neurotransmitters upon exposure to various stress agents. A shift in the brain perception and processing following any stimulus results in false or hyperfiring of the neurons, which results in anxious behaviour in affected individuals. However, studies have reported that a misbalance in the cellular homeostasis of the brain also results in such behavioural deficits, which opens up a whole dimension of psychotic studies. In molecular aspects, long-term exposure to stressors results in astrogliosis, further upregulating the expression of GFAP genes. 94 Microglial activation is also reported in brain samples of attention-deficit/hyperactivity disorder (ADHD). These patients are observed to have low dopamine D1 receptor density in the dorsolateral prefrontal cortex, which is reported to be associated with deficits in attention and information processing speed. Microglial activation causes neuroinflammation and is responsible for hyperactivity and depression-like symptoms. Again, age-dependent decrease in immunoreactive astrocytes in the prefrontal cortex is reported to cause changes in glutamate/GABA neurotransmission, and such changes, along with a decrease in oligodendrocyte population in prefrontal cortex and amygdala, are linked to depression-like behaviour.92, 94, 96 Postmortem findings also correlate mood disorder with glial pathology, and subjects with mood disorders and severe depressive bipolar disorders are reported to have elevated levels of S100β protein in serum. S100β is released by astrocytes, and its serum elevation points towards astrogliosis in affected individuals. 97 In Alzheimer’s brain as well, the astrocytes remain highly activated with a significant increase in reactive astrocyte population along with a low oligodendrocyte count. Similarly, in schizophrenic brain also, gliosis is prominent with increased density of reactive astrocytes along with reduced oligodendrocytes. Such alterations in astrocytes and oligodendrocytes, observed in schizophrenic conditions, are due to changes in a set of genes involved in DNA, RNA, protein, lipid and carbohydrate metabolism in astrocytes and oligodendrocytes.95, 97 Furthermore, neuronal loss or cytoarchitectural damage is mainly responsible for cognitive impairment like learning and memory loss, and such cognitive impairment is also seen in schizophrenic patients. Thus, unhealthy neuronal and glial conditions are related to behavioural and cognitive loss, which, further in chronic situations, give rise to neurological disorders like schizophrenia.
Multiple Exposure to Early-life Stress and Development of Schizophrenia
Schizophrenia, being a psychotic disorder, is triggered by more than one stress factor, and the chances of development of this disorder increase by many folds when all the above-mentioned stressors act cumulatively. Although there are limited studies which talk about the cumulative impact of multiple early-life stressors, some of the studies have reported that viral infection, along with social stress like loneliness and fear of the future, could impact an individual, resulting in a spike in schizophrenia-like symptoms. Scientists have also reviewed the multi-hit concept of schizophrenia and reported that schizophrenia is related to multiple risk factors. Although more research is needed in this area, the relation between the multi-hit hypothesis and schizophrenia is well established to be carried as an emerging area of research.95, 98
Conclusion
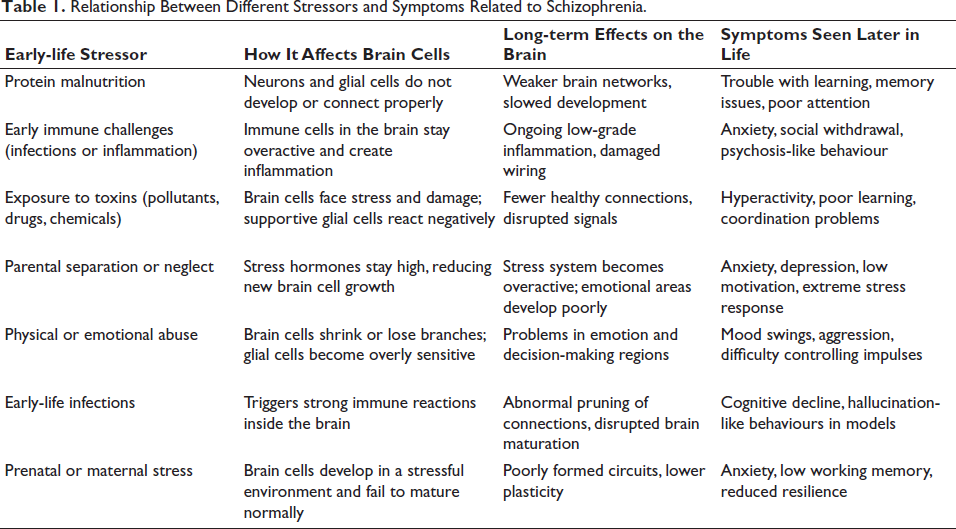
The developing brain is vulnerable, and any adversity, such as maternal malnutrition, immunologic episodes, exposure to toxicants, and even parental separation and abuse, negatively influence the developing brain. It is now evident that such challenges affect both formation and functional maturation of the astrocytes, oligodendrocytes and microglia from their respective precursors and thus negatively challenge the functional maturation of the brain cytoarchitecture. The neurons are equally affected. Such early-life stressors via the glia leave a long-term impact on the neurons, altering cognitive and behavioural abilities in the nervous system, potentially causing schizophrenia (Table 1). Thus, a healthy and stress-free environment, mainly during the crucial perinatal age, is necessary for the proper functioning of glial cells, which otherwise shall influence neuronal function, resulting in abnormalities that can cause learning impairment, memory loss, anxiety and hyperactivity-like disorder, depression, and so on, contributing towards the development of neuropsychiatric disorders like schizophrenia.
Relationship Between Different Stressors and Symptoms Related to Schizophrenia.
Footnotes
Authors’ Contributions
IKP, TS and NP conceptualised. TS wrote and formatted the manuscript. IKP and NP modified and reviewed the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical Approval and Informed Consent
Not applicable.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Indian Council of Medical Research, Government of India, New Delhi, ICMR-Emeritus Scientist grant (No. HRD/IES-2025/4, dated 08.04.2025) to IKP and Department of Biotechnology, Government of India, Research Associate grant (DBT-RA/2023IJanuary/NE/3421) to TS are thankfully acknowledged.