
Abstract
Johanson–Blizzard’s syndrome (JBS) is a rare cause of chronic diarrhoea in infancy. It is an autosomal recessive disease characterized by exocrine pancreatic insufficiency, multiple characteristic dysmorphic features, multisystem involvement and developmental delay. JBS is a very rare disease with roughly about 70 cases reported in the literature. We are reporting this case of a male infant who presented with chronic diarrhoea and had typical facial features along with café-au-lait spots, and the whole exome sequencing revealed a novel homozygous UBR1 mutation.
Keywords
Introduction
Johanson–Blizzard’s syndrome (JBS) is a rare genetic disorder with autosomal recessive inheritance. There is a loss-of-function mutation in the UBR1 (ubiquitin-protein ligase E3 component N-recognin 1) gene, which is located on chromosome 15q15-2. JBS was first described by Ann Johanson and Robert Blizzard in 1971. 1 It is a very rare genetic disease, and the frequency is estimated to be 1 in 250,000 births. 2 Only about 70 cases were reported in the literature.
Clinical presentation is very variable and can differ significantly from one patient to another. The most characteristic feature is exocrine pancreatic insufficiency. Other clinical features include dysmorphic facial features with a small beaked nose, dental abnormalities, hypothyroidism, failure to thrive and mental retardation. 3 The disease-causing locus is mapped to chromosome 15q15-21 with an identified mutation in the UBR1 gene which encodes ubiquitin ligase. Here, we report a 45-day-old male infant who was primarily referred due to severe chronic infantile diarrhoea and found to have classical features suggestive of JBS, along with café-au-lait spots, which is a relatively uncommon finding. In addition, whole exome sequencing revealed a new mutation in the UBR1 gene.
Case Report
A 45-day-old male infant was referred to our centre because of persistent diarrhoea and failure to thrive. There were no similar complaints among family members, and the previous sibling was healthy.
The infant was born at full term to a non-consanguineous couple and was small for gestational age with a birth weight of 1,800 g. His weight, length and head circumference were less than third centile. He was exclusively breastfed and remained well till the second week of life. In the third week, he developed frequent, loose stools. He was managed in the referral hospital with a low-lactose formula. He was also given a packed cell transfusion due to anaemia. As the infant continued to have diarrhoea, he was referred to our centre for further management.
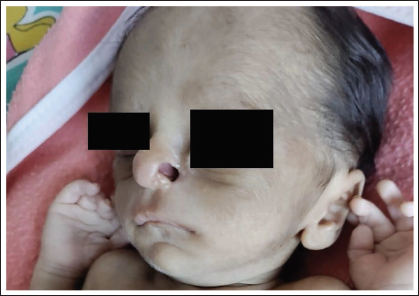
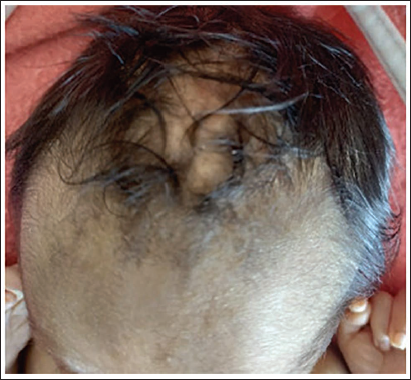
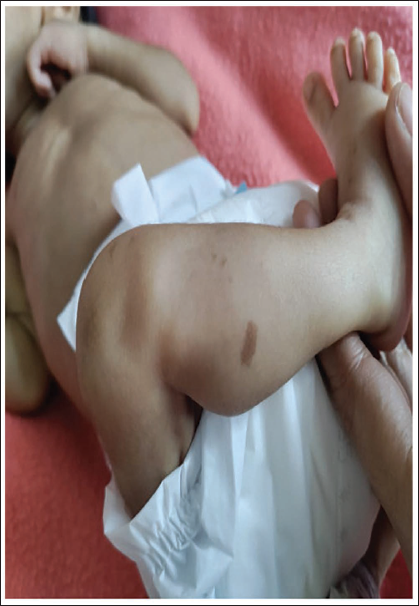
At the time of admission, the infant was pale and lethargic. On physical examination, he had dysmorphic facies with mongoloid slant, low-set ears, absent ala nasi, breaking of the nose and cutis aplasia over the scalp (Figures 1 and 2). Multiple café-au-lait spots were also present over the legs and perineum (Figure 3). Septic workup done at admission showed a C-reactive protein of 29 mg/dL. Complete blood picture showed haemoglobin of 8 and normocytic, normochromic anaemia. As the infant received a blood transfusion prior to referral, further investigations to look for the cause of anaemia could not be done. Complete urine examination was normal. He was started on intravenous piperacillin/tazobactam and amikacin after sending blood and urine cultures which were sterile at 48 hours. Hence antibiotics were stopped. The stools were large and watery with a foul smell. The stool routine was normal and was negative for reducing substances. Due to persistent loose stools despite being on a low-lactose formula, an extensively hydrolyzed formula was started.
Beaked nose, hypoplastic ala nasi
Cutis aplasia
Café-au-lait spots
As physical features were suggestive of JBS, paediatric gastroenterologist consultation was sought, and investigations were done to look for exocrine pancreatic insufficiency, which showed reduced levels of serum amylase of 6.8 U/L and serum lipase of 2 U/L. The faecal elastase-1 level was 55 µg/g of stool. Faecal elastase-1 < 200 µg/g of stool is indicative of exocrine pancreatic insufficiency. Hence, the infant was started on pancreatic enzyme supplements, multivitamins and oral iron supplementation. The hearing screen (automated auditory brainstem response) showed bilateral sensorineural hearing loss. T3, T4, free T4 and thyroid-stimulating hormone levels were normal.
He had improvement in symptoms with a decrease in the frequency of stools and normal stool consistency after starting on extensively hydrolyzed formula and pancreatic enzyme supplements. He had consistent weight gain. He was discharged after 10 days of hospital stay. He was gaining weight in follow-up but lost to follow-up 1-month post-discharge.
Genetic Studies
Whole exome sequencing was done after genetic counselling, and it showed homozygous missense variation in exon 17 of the UBR1 gene that results in amino acid substitution of lysine for glutamic acid at codon 647. It is classified as a variant of uncertain significance. This variant has not been reported in the literature so far. Genetic testing was advised to both parents to look for carrier state, but they could not get it done due to financial constraints.
Discussion
JBS is a rare disorder exhibiting various genetic abnormalities with an incidence of around 1 per 250,000. It is autosomal recessive in inheritance and is more prevalent in consanguineous parents. In our case, there was no history of consanguinity.
The infant presented here was symmetric intrauterine growth restriction (IUGR) with failure to thrive and typical facial features. Symmetric IUGR in this infant is due to intrinsic growth failure similar to that seen in other syndromic infants. He also had scalp defects and hypotonia. Café-au-lait spots in JBS have been described in only a few previous case reports of JBS. He had exocrine pancreatic insufficiency, which is pathognomonic for JBS. However, this infant did not present with steatorrhoea, which is the characteristic symptom of exocrine pancreatic insufficiency, but had large watery stools. In neonates and infants, steatorrhoea can be less obvious, as the only source of dietary fat is milk fat which is liquid at body temperature and solid at room temperature, and the dietary carbohydrates (lactose) and the milk proteins (casein and whey) cannot form a solid complex with fats. Stools can also be watery because of the osmotic load received by the intestine. 4 In addition, there is a history of anaemia, and he received one blood transfusion before referral to our hospital. There are very few JBS cases with anaemia as one of the features like in our case, reported in the literature. In infants, nutritional anaemia is unusual before 6 months of age. However, the nutritional consequences of malabsorption can appear early in some cases due to factors such as low birth weight, hypothyroidism reduced red cell production and survival. 5 Exocrine pancreatic insufficiency with anaemia is also seen in Shwachman–Diamond’s syndrome (SDS) and is the second most common inherited syndrome causing exocrine pancreatic insufficiency after cystic fibrosis. SDS needs to be ruled out in any infant presenting with anaemia with exocrine pancreatic insufficiency. SDS is characterized by multisystem involvement, and infants often have other skeletal abnormalities such as short stature with or without metaphyseal dysplasia and rib cage abnormalities. Neutropenia is the most common haematological abnormality which is not seen in this infant and anaemia in SDS can be macrocytic or normocytic. 6
The genetic basis of JBS was found to be due to mutations in the UBR1 gene, located on chromosome 15q15-q21.1. UBR1 gene encodes ubiquitin ligase of the N-end rule pathway, a ubiquitin-dependent proteolytic pathway, and is considered to play a critical role in the development and maintenance of acinar cells. In patients with JBS, destruction of acinar tissue of the pancreas may begin in utero and hence can lead to the development of exocrine pancreatic insufficiency.2, 7 In the earlier reported cases, truncating and splicing mutations in UBR1 were described as the most common mutations followed by missense mutations. 7 Later cases reported small in-frame deletions and compound heterozygous mutations in the UBR1 gene. 8 In the index case, whole exome sequencing revealed a novel homozygous missense mutation in exon 17 of the UBR1gene, which has not been reported previously.
Infants with JBS should be screened for other anomalies such as renal anomalies, dental anomalies, hypothyroidism and diabetes and provided with appropriate genetic counselling. Therefore, optimal management of these infants requires a multidisciplinary team involving a paediatrician/neonatologist, paediatric gastroenterologist, endocrinologist, dentist, geneticist and physiotherapist. They also need long-term care and follow-up for the management of pancreatic insufficiency and hypothyroidism, treatment of frequent respiratory infections, management of hearing loss, physiotherapy and educational rehabilitation.
Conclusion
JBS is a rare autosomal recessive disorder affecting multiple organ systems of the body, and hence, multidisciplinary team involvement is important in managing these cases even during follow-up. In the index case, the infant had characteristic facial features and failure to thrive with pancreatic insufficiency, which led to suspicion of this syndrome, later confirmed by molecular studies. Whole exome sequencing in this infant revealed a homozygous missense variation in exon 17 of the UBR1 gene which has not been reported in the literature so far.
Paediatricians need to consider molecular testing of the UBR1 gene not only for the confirmation of the diagnosis in the affected child but also for confirming carrier status in both parents and to offer appropriate counselling to the family so that prenatal diagnosis can be offered for future pregnancies.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Approval was obtained from the Ankura Ethics Committee for biomedical research.
Funding
The authors received no financial support for the research, author ship, and/or publication of this article.
Informed Consent
Written informed consent for publication was obtained from the infant’s father.