
Abstract
We report on a triplet pregnancy of consanguineous parents with one fetus being affected by recurrent Johanson-Blizzard syndrome (JBS). At autopsy in the 35th gestational week, the affected triplet presented with an especially severe and lethal manifestation of the disorder as compared to his elder affected brother and to cases in the literature, thus exemplifying great interfamilial and intrafamilial phenotypic variability. Arhinencephaly and cystic renal dysplasia associated with urethral obstruction sequence were features not described previously in the literature. In addition to the lack of exocrine acini as the characteristic feature of JBS, the pancreas revealed a resorptive inflammatory reaction with infiltration by eosinophilic granulocytes that focally dispersed onto islets of Langerhans, thus favoring a progressive destructive rather than primary dysplastic process and possibly explaining the occurrence of diabetes mellitus in later life. JBS maps to chromosome 15q15-q21.1 and is associated with mutations in the UBR1 gene. Testing the fetus and the affected sibling revealed a homozygous truncating mutation in UBR1. The resulting absence of the UBR1 protein was confirmed by Western blot. Immunohistochemical staining using a commercial anti-UBR1 antibody demonstrated staining, presumably artifactual. This finding suggests that, until an appropriately validated antibody has been identified, this modality should not be utilized for diagnosis or confirmation of this disorder.
Keywords
INTRODUCTION
Johanson-Blizzard syndrome (JBS) (OMIM #243800) is a rare, autosomal-recessive, multiple-malformation syndrome. Striking features are ectodermal dysplasia with midline scalp defects, abnormal hair pattern, and absent permanent teeth; a characteristic facial appearance that includes hypoplastic or aplastic nasal alae, midfacial hypoplasia, a long upper lip with downturned corners of the mouth, and microgenia; sensorineural hearing loss; hypothyroidism; occasionally hypopituitarism; and obligatory exocrine pancreatic insufficiency. Severe growth retardation is attributed to malabsorption and lack of growth hormone. Further anomalies, eg, of the intestinal or genitourinary tract and of the heart, including cardiomyopathy, may be associated. Mental retardation is not obligatory but may occur to varying degrees [1–10].
In 2005 it was shown that deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, representing a proteolytic pathway of the ubiquitin system and encoded by the UBR1 gene in 15q15-q21.1, causes JBS [11]. Subsequently, UBR1 mutations have been demonstrated in additional JBS families [9,10,12,13].
To date, there are few prenatal reports of JBS [14,15]. Here we present a triplet pregnancy of which one fetus had recurrent JBS with especially severe and lethal manifestations of the disorder and discuss the autopsy, molecular, and immunhistochemical findings.
METHODS
Clinical examinations, autopsy, molecular studies, and (immuno-)histochemistry
The affected elder son (case 1) was examined clinically. Autopsy was performed on the deceased affected triplet (case 2).
Molecular genetic studies were performed after informed consent was obtained from the parents. Genomic DNA from living family members and the deceased affected child was extracted from blood samples and a frozen tissue sample, respectively, according to standard procedures. All 47 known coding exons, including the flanking intronic regions of the UBR1 gene, were amplified by polymerase chain reaction (PCR). PCR amplicons were purified and subjected to direct sequencing using the Big Dye Terminator Cycle Sequencing Kit v. 3.1 (Applied Biosystems, Darmstadt, Germany) and an automated sequencer (ABI3730, Applied Biosystems) for detection. Sequences were compared to the reference sequences deposited in the public database (NM_174916). Oligonucleotide primer sequences and PCR protocols are available on request.
For immunohistochemical investigations of the pancreas in case 2, paraffin-embedded specimens were processed and incubated with trypsin-(rabbit polyclonal), insulin-(mouse monoclonal), and glucagon-(rabbit polyclonal) antibodies (Calbiochem, Darmstadt, Germany; Leica Microsystems, Wetzlat, Germany; Dako, Hamburg, Germany), as well as somatostatin-(rabbit polyclonal) antibody (Dako) using standard techniques and the Bond maX automated immunostainer (Leica Microsystems). In addition, immunohistochemistry for ubiquitin (rabbit polyclonal antibody, dilution 1:1000, Dako) and UBR1 (mouse monoclonal antibody, clone 2F5, dilution 1:500, Abnova, Taipei, Taiwan) was performed. Antigen retrieval was performed with a steamer for 30 minutes in DIVA buffer (no. DV 2004MX, Biocare Medical). The staining procedure was performed in a Dako Autostainer. Bound antibodies were visualized by the Dako REAL EnVision kit (Dako no. K5007) containing the DAB chromogen.
RESULTS
Case 1 (affected sibling 1)
Case 1, a 2-year-old boy, is the product of the 2nd pregnancy of a 27-year-old woman. The woman and her husband were of Kurdish origin and were 2nd cousins. They were both healthy, as was their eldest child, a daughter. The family history was negative for congenital malformations or mental retardation.
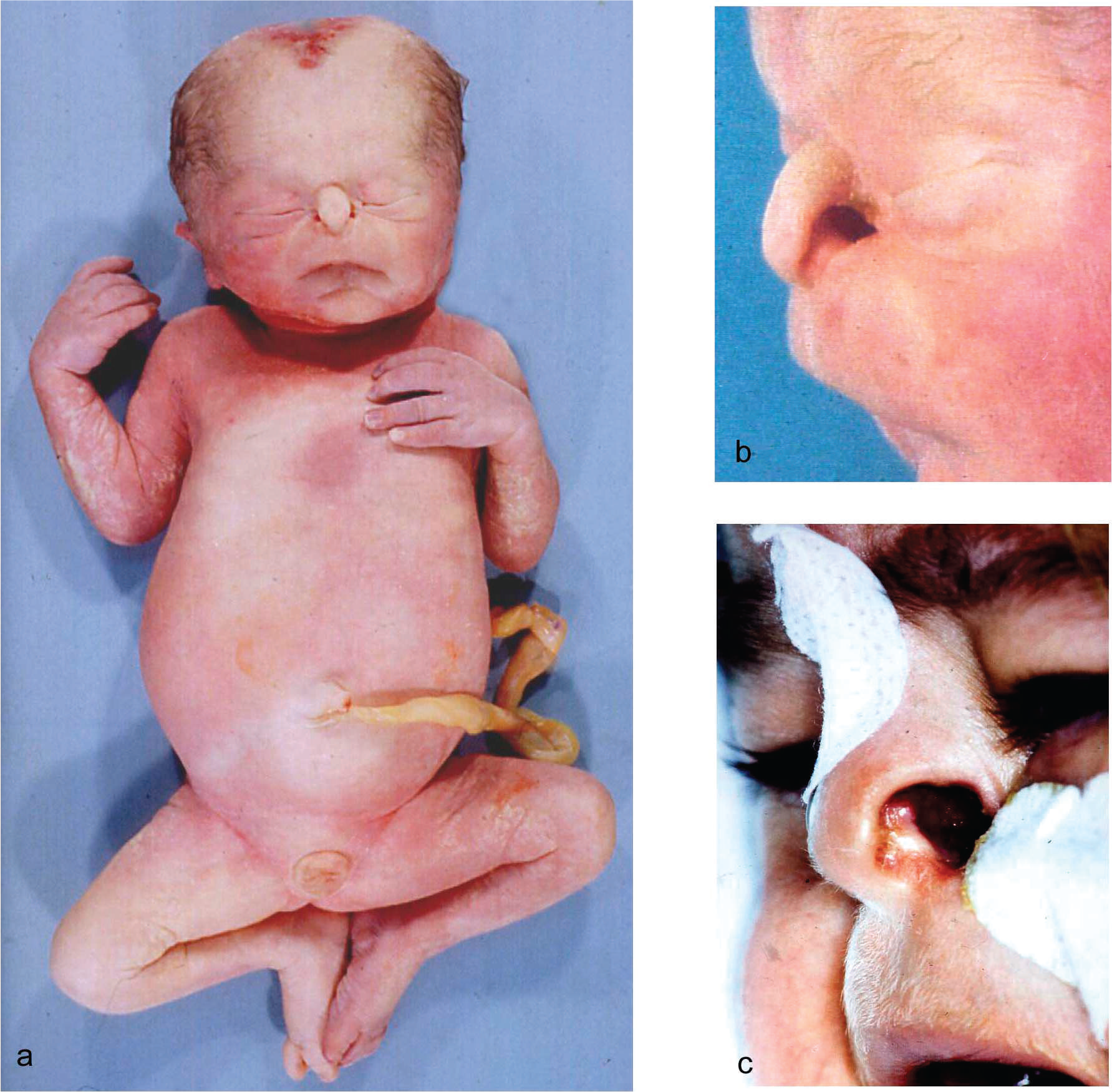
JBS had been diagnosed after birth in the 2-year-old boy, who was born at term with Apgar scores of 9/9/10, a birth weight of 3020 g, a body length of 52 cm, and a head circumference of 35 cm. Diagnosis was based on the clinical findings of scalp defects, hypoplastic nasal alae, imperforate anus, and hypoplastic genitalia with perineal hypospadias and unilateral cryptorchidism (Figs. 1,2). Mechanical ventilation was necessary for 4 weeks because of respiratory failure. Colostomy was performed. The further clinical course was complicated by a severe pancreatic insufficiency with malabsorption, voluminous feces, and severe growth retardation. Substitution with pancreatic enzymes led to some gain of weight, which was nevertheless always far below the 3rd percentile. Repeated pulmonary infections resulted in restrictive pneumopathy with a honeycomb appearance on lung biopsy. At the age of 12 months, tertiary/hypothalamic hypothyroidism was diagnosed. The patient had low concentrations of prolactin and no growth hormone response to an arginine tolerance test. By the age of 2 years, the patient was unable to sit or to crawl and was found to be deaf, with no speech development.
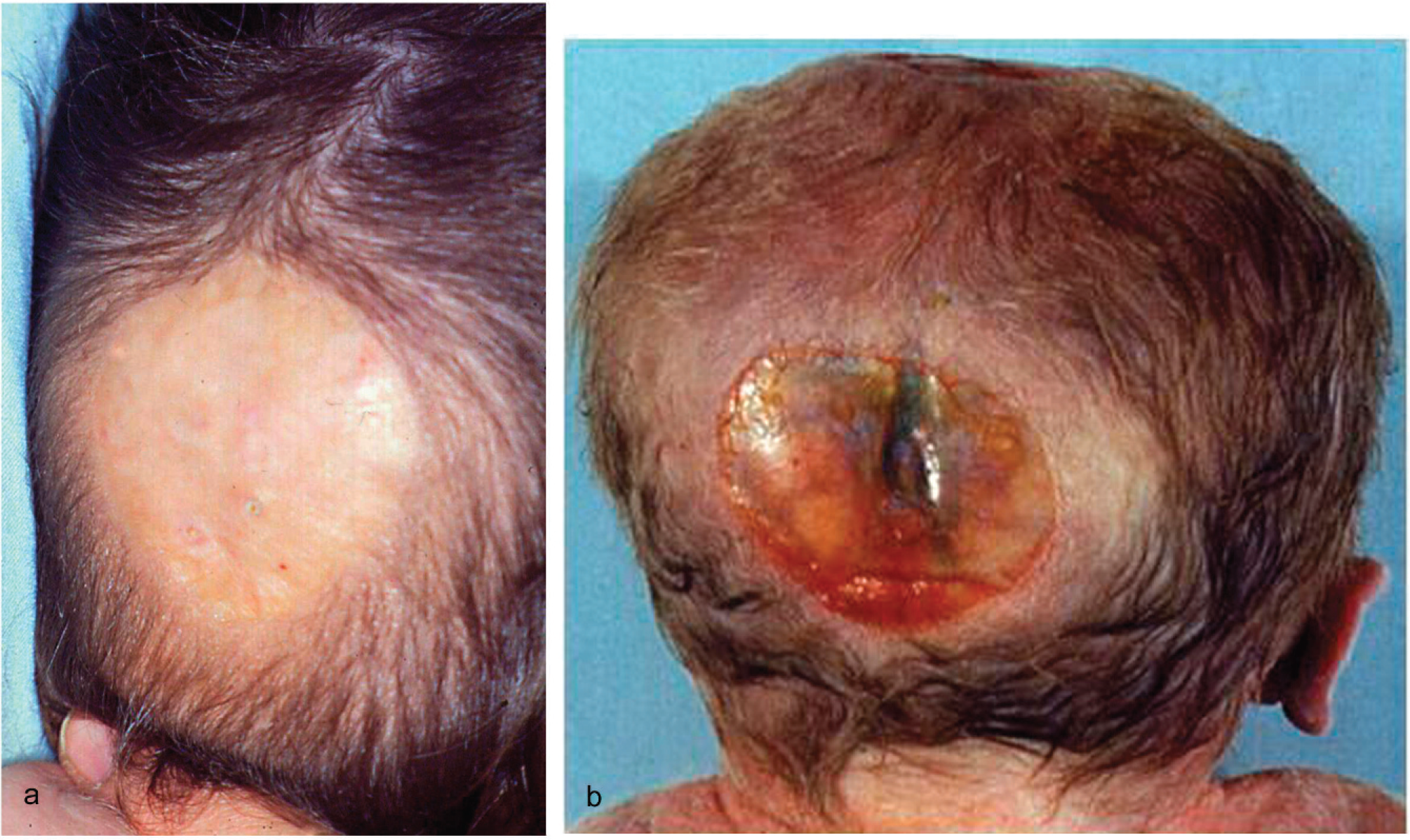
Occipital scalp defects in case 1 (
Case 2 (affected sibling 2, fetus)
Case 2 is the product of the same couple's 3rd pregnancy. Ultrasound examination at 17 weeks showed a triamniotic, trichorionic triplet pregnancy. The 3rd triplet displayed oligohydramnios, growth retardation, bilateral hydronephrosis with a dilated urinary bladder, a normal male 46,XY karyotype, and an amniotic alpha fetoprotein (AFP) value that had increased to a multiple of the median of 9.5 (normal range, 0.5–2.0 multiples of the median). Growth retardation in the 3rd male triplet increased to 2 weeks. At 33 weeks’ gestation he developed cardiomegaly with pericardial effusion. Caesarean section was performed at 35 weeks. The 1st two triplets delivered were a healthy female and male. Apgar scores of the 3rd triplet (ie, case 2) were 1/1/1. The boy died 40 minutes after delivery as a result of respiratory insufficiency.
The deceased male triplet had a birth weight of 2080 g, a length of 43 cm, and a head circumference of 31 cm. Multiple congenital malformations were documented at autopsy and included brachycephaly; a high forehead; large frontoparietal and occipital scalp defects of 4 × 4.5 and 4 × 5.5 cm, respectively; interorbital placement of a short flat nose with aplastic nasal alae, beaked nasal tip, and extremely short nasal bridge; distinct hypotelorism; upslanting palpebral fissures; mild ocular proptosis; epicanthic folds; missing eyebrows with temporal hair tongues; a hypoplastic midface; a smooth, long philtrum; a thin upper lip; downturned corners of the mouth; a sublabial groove; micro- and retrognathia; large ears; and underdeveloped nipples (Figs. 1,2). There was arhinencephaly with bilateral absence of the olfactory bulbs and tracts. The thyroid gland was hypoplastic with large colloid-filled follicles. The heart was hypertrophic. There were pleural and pericardial effusions and ascites and distinct hypoplasia of the lungs as a result of oligohydramnios. Abdominal distension was secondary to excessively enlarged kidneys and to megacystis in line with a urethral obstruction (prune-belly) sequence. The right kidney displayed cystic dysplasia (Potter type 2) with densely packed cysts on the cut surface; histology revealed abortive formation of glomerula and a complete absence of intact nephrons. The left kidney was moderately enlarged and of the obstructive cystic dysplastic type (Potter type 4), showing one to two layers of cysts in subcapsular cortical regions with otherwise normal renal tissue differentiation. Dilated renal pelvices, extremely dilated kinked ureters, and dilatation of urinary bladder and proximal urethra were secondary to urethral obstruction. This was caused by urethral compression through a urethral diverticle at the level of an absent prostate and by a cryptic micropenis that was hidden in the depth of the subpubic soft tissues and displaying glandular epispadia (Figs. 3,4). Cryptorchidism and a patent anus in normal position were also present. The malformation pattern confirmed recurrence of JBS.
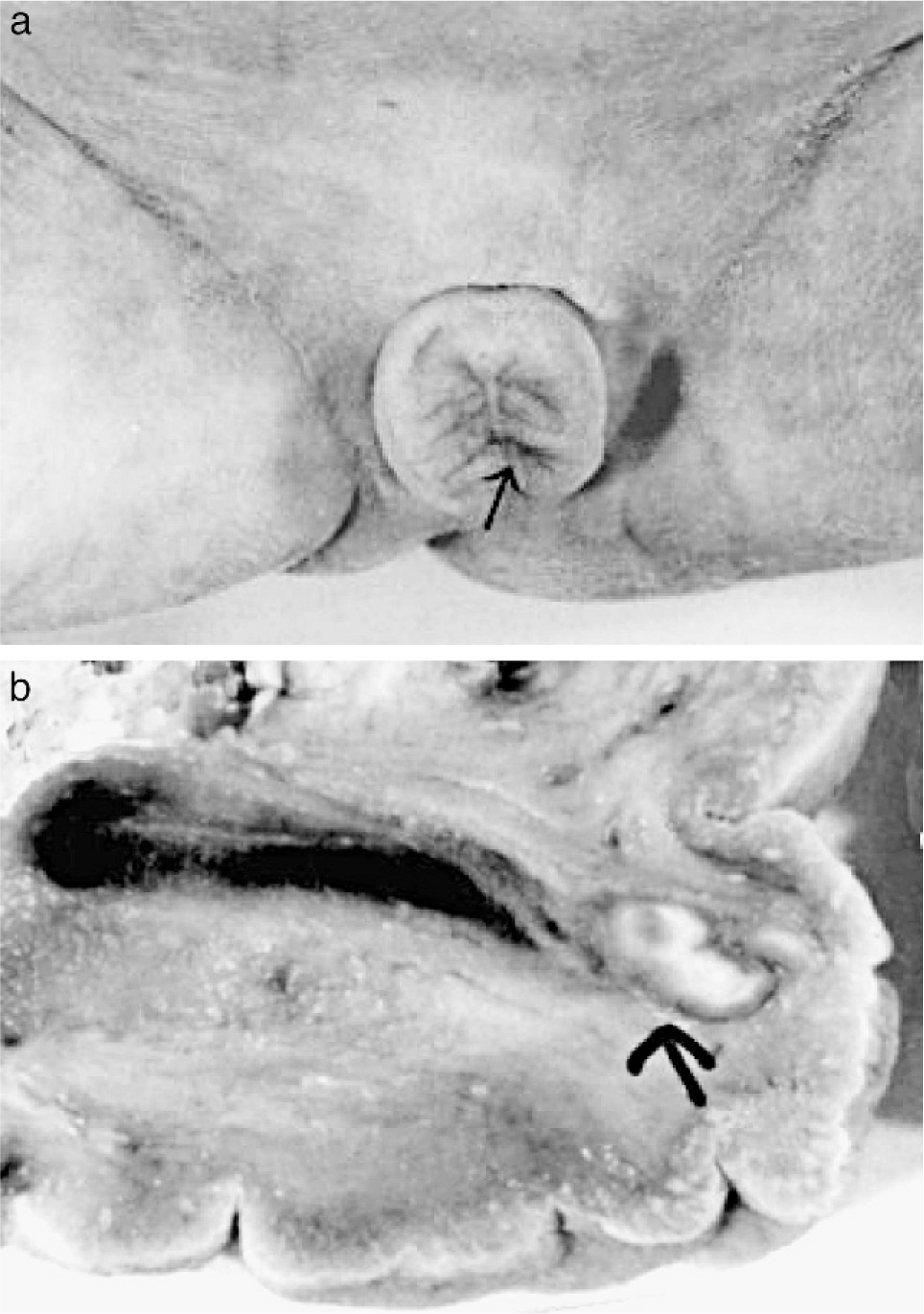
Case 2.
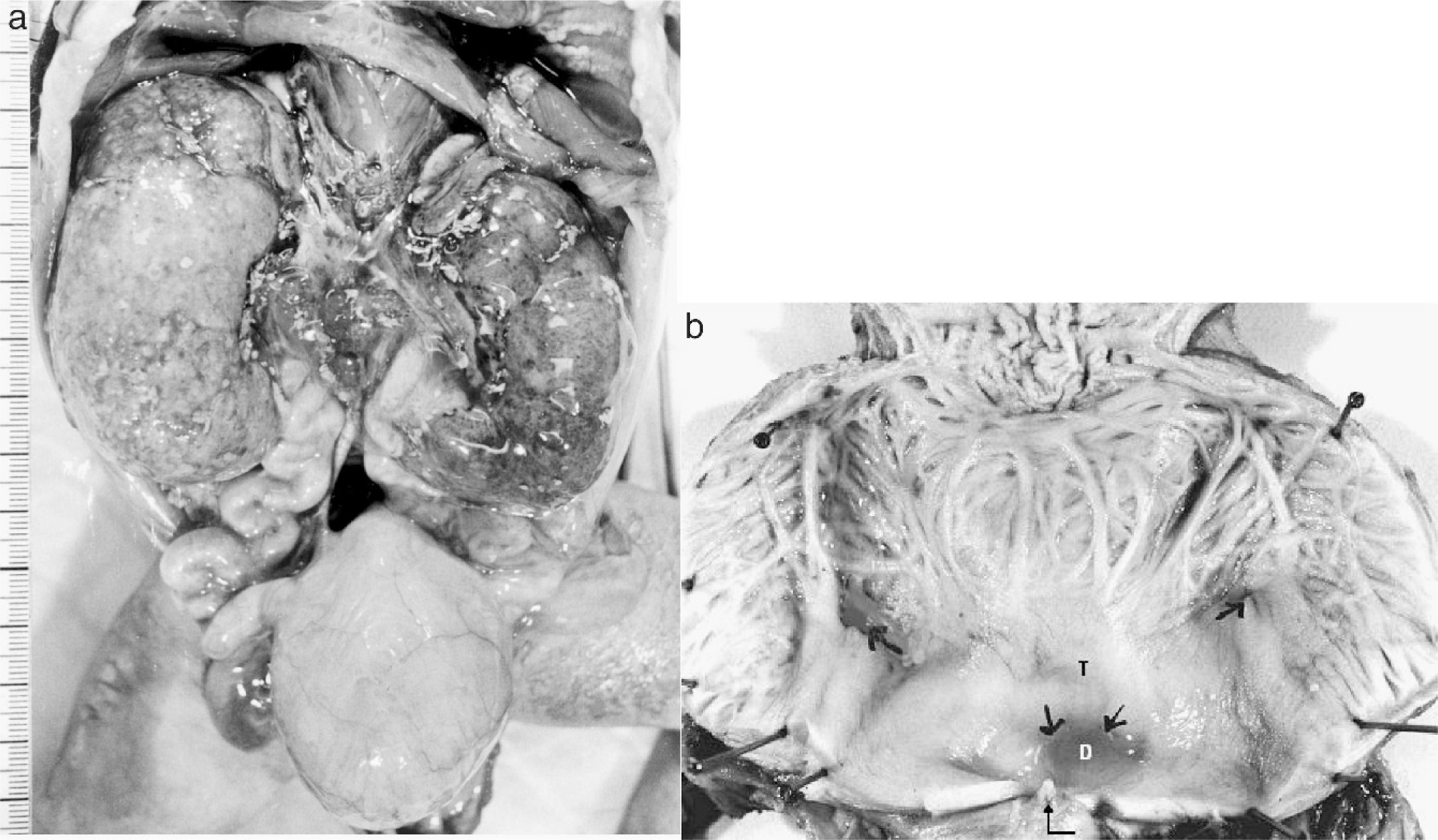
Intra-abdominal aspect of urinary system in case 2 showing dilated bladder (H), dilated and kinked ureters, enlarged cystic kidneys (
Molecular genetic and Western blot studies in cases 1 and 2
The fetus and his affected elder brother, were found to share a region of autozygosity, including the UBR gene locus, and a homozygous mutation c.1759C>T was identified in exon 15 of UBR1. This mutation predicts a premature translational stop codon (p.Q587X) and was demonstrated to result in a complete lack of UBR1 protein expression by Western blot using a noncommercial UBR1 antibody [11,16–18]. The consanguineous parents were shown to be heterozygous carriers of the same UBR1 mutation.
Immunohistochemistry of the pancreas of case 2 revealed that the pancreas was made up of a few small lobules with excretory ducts, abundant islets of Langerhans, and a nearly complete absence of exocrine acini. These were replaced by connective tissue that was focally infiltrated by masses of eosinophilic granulocytes encroaching upon and marginally destroying the islets of Langerhans. Immunohistochemical studies using trypsin, insulin, glucagon, somatostatin, and ubiquitin antibodies evidenced an almost complete absence of trypsin, with little expression within the scarce residual acini. Insulin, glucagon, somatostatin, and ubiquitin expression in islets was almost normal compared to age-matched controls, with the exception of inflammatory or centrally necrotic islets displaying a distinct reduction in insulin expression (Fig. 5). Immunohistochemical investigations using a commercially available monoclonal antibody against UBR1 protein on paraffin sections of pancreatic tissue after antigen retrieval did not reveal significant staining differences between the affected fetus and a control fetus, suggesting that this particular antibody does not reliably recognize UBR1 protein in paraffin-embedded material.

Pancreatic tissue of case 2 displaying absence of exocrine acini but abundant islets of Langerhans (
DISCUSSION
Diagnosis of JBS is based primarily on clinical criteria. Its most striking external features are scalp defects and hypoplastic or aplastic nasal alae, which allow a preliminary diagnosis in severely affected persons [6]. In combination with other common morphological and clinical abnormalities, such as growth retardation and malabsorption owing to exocrine pancreatic insufficiency, the diagnosis can be made with certainty if the variable expression of features is considered.
The gene responsible for JBS was identified in 2005 by Zenker and colleagues [11]. UBR1 spans over 161 base pairs and contains 47 exons that code for ubiquitin-protein ligase E3 component N-recognin1 (UBR1) with a molecular weight of 200 kDa. UBR1 is broadly expressed, showing its highest levels in skeletal muscle, kidney, and pancreas [11,17]. Several mutations scattered across the entire gene have been described. They mainly represent nonsense, frame shift, and splice-site mutations predicting a premature truncation of the UBR1 protein. In a few cases, missense mutations or in-frame deletions allow some residual UBR1 function and are responsible for mild manifestation of JBS, eg, in compound heterozygotes [9,10,12,13,18]. Our two cases presented with a homozygous nonsense mutation in exon 15 that resulted in complete loss of UBR1 expression in the Western blot [11]. This coincides well with the severe phenotype of our case 2. However, in immunohistochemical studies of the fetal pancreas using a commercial UBR1 antibody, UBR1 immunolocalization was comparable to that of an age-matched control. This indicates that the commercial UBR1 antibody—primarily developed for Western blot investigations—does not reliably function on paraffin-embedded material.
To date, 2 reported cases have been diagnosed prenatally [14,15]. The authors recognized the syndrome through ultrasound at 21 weeks’ gestation by the hypoplastic alae nasi and a dilated colon. In our case of a triplet pregnancy, we were aware of a prior risk of 25% for JBS.
Since hydronephrosis, mentioned by Johanson and Blizzard [6] in their original report, was detected in one fetus by ultrasound, we suspected that this fetus was affected by the disorder. Because of his intrauterine position and oligohydramnios, the facial features could not be assessed in detail. However, strongly increased amniotic AFP levels, known to be related to scalp defects, strengthened the suspicion of recurrent JBS.
Autopsy findings confirmed the diagnosis of JBS. In addition to the characteristic external features (Figs. 1,2), arhinencephaly was found. Arhinencephaly or resulting anosmia had not been mentioned in former reports, but fits well with the other facial midline defects, such as the hypotelorism and the intraorbital placement of a small nose. The association of arhinencephaly and an almost ethmocephalic face may be regarded as part of the holoprosencephaly spectrum.
Dilatation of the urinary bladder and proximal urethra, oligohydramnios, and lung hypoplasia are part of an early urethral obstruction sequence (prune-belly sequence). However, the cystic renal dysplasia, the extremely kinked ureters, the absent prostate, and the cryptic micropenis with glandular epispadia (Figs. 3,4) may rather be regarded as a complex urogenital developmental field defect. Urogenital anomalies, including milder anomalies, such as enlargement of the renal pelvis, renal microcysts, bicornuate uterus, septate vagina, cryptorchidism, micropenis, and hypospadias, have been mentioned in 25% of JBS cases [1,4,5,19,20], but never to the extent as seen in our case 2, where urogenital malformations were responsible for early postnatal death. In 47% of cases with JBS, anorectal anomalies such as anal atresia or ectopia are common findings [6,7]. Anal atresia may occur with rectovaginal fistula in females with JBS.
Absence or paucity of exocrine pancreatic tissue explains malabsorption in children with JBS. It has been described after autopsy as “total absence of acini in the presence of ductuloinsular complexes with clusters of islets of Langerhans and mildly dilated and metaplastic intralobular ducts” [4,21] and as “lacking exocrine tissue being replaced by connective tissue with focally prominent ‘haematopoetic’ cells” [5]. Because of these morphologic changes, Gould and colleagues [5] suggested a primary pancreatic dysplasia but could not rule out early acinar destruction. The pancreatic infiltration by eosinophilic granulocytes in our case 2, which demonstrated a resorptive inflammatory reaction, may be in favor of a destructive rather than a primary dysplastic process. When it has spread to the islets of Langerhans (Fig. 5), it may explain the progression of pancreatic involvement with diabetes mellitus in later life. Diabetes mellitus has been described in 11- and 19-year-old girls with JBS [22–24].
Case 1 presented with additional restrictive pulmonary disease of unknown cause. Our observation of unexplained ventilator dependency in a term newborn with later recognized honeycomb lung (case 1) and of congenital bronchiectasis in the deceased sibling fetus (case 2) indicates that this organ may be affected before birth. This is in line with previous reports suggesting that chronic lung disease may be part of JBS [19,25].
Hypothyroidism with typical histological changes, namely atrophy and dilated colloid-filled follicles, is a common finding in JBS [1,4–7,21,26,27]. It was demonstrated in our case 2 at autopsy. Hypopituitarism has also been reported in a few instances [25–29]. In our case 1, no additional pituitary function testing or nuclear magnetic resonance tomography was performed. However, low concentrations of prolactin and growth hormone and tertiary hypothyroidism suggest a complex hypothalamic/hypophyseal insufficiency, supporting the hypothesis that hypopituitarism can be part of the spectrum of JBS [26,29]. In association with arhinencephaly, hypothalamic/hypophyseal insufficiency could be explained by similar pathogenetic pathways as are suggested for the combination of anosmia and hypothalamic hypogonadism in Kallmann syndrome [30].
The affected siblings presented here demonstrate the great intrafamilial variability of expression in JBS and suggest the possibility of prenatal detection by elevated amniotic AFP levels. One case manifested especially severe urogenital anomalies resulting in renal insufficiency and lethal urethral obstruction sequence. Such cases have an adverse effect on fetal viability and thus typically escape recognition in JBS.
Footnotes
ACKNOWLEDGMENTS
We are very grateful to Dr Ernestine Piecha-Ngo (Marburg) for the photographic documentation of case 2 and to Marianne Kolitsch (Marburg) and B. Beerwirth (Münster) for their technical help.