
Abstract
This study reports three patients with Cat-eye Syndrome (CES), two of which present a previous clinical diagnosis of Craniofacial microsomia (CFM). Chromosomal microarray analysis (CMA) revealed a tetrasomy of 1,7 Mb at the 22q11.2q11.21 region, which is the typical region triplicated in the CES, in all patients. The most frequent craniofacial features found in individuals with CFM and CES are preauricular tags and/or pits and mandibular hypoplasia. We reinforce that the candidate genes for CFM features, particularly ear malformation, preauricular tags/pits, and facial asymmetry, can be in the proximal region of the 22q11.2 region.
Keywords
Introduction
Craniofacial Microsomia (CFM) (OMIM 164210), also known as Oculoauriculovertebral Spectrum (OAVS) and Goldenhar syndrome, is a rare condition caused by a morphogenesis defect of the first and second pharyngeal arches. All structures derived from these arches, such as the ears, eyes, muscles, facial bones, and cervical vertebrae, may be affected resulting in a large spectrum of clinical features. 1
Additionally, many of these features overlap with other syndromes related to the development of pharyngeal arches and should be considered in the differential diagnosis of CFM, such as Cat-Eye syndrome (CES) (OMIM # 115470), Treacher Collins syndrome (OMIM # 154500, # 248390, # 613717, # 61839), Auriculocondylar syndrome (OMIM # 602483), Bixler syndrome (OMIM # 239800), Hemifacial myohyperplasia sequence (OMIM 606773), Branchiootorenal (BOR) syndrome (OMIM # 113650, # 610896, CHARGE syndrome (OMIM # 214800), Mandibulofacial dysostosis (OMIM # 610536), 2 and TP63-Related Disorders (OMIM # 603273) 3
Multifactorial inheritance has been proposed as the best model in the majority of cases. Maternal diabetes and gestational exposure to retinoic acid and vasoactive drugs are among the most reported environmental factors. The use of reproductive technology and smoking during the second trimester have also been identified as risk factors in more recent studies. 4 Regarding genetic causes, copy number variations (CNV) at 1p22.2, 1p31.1, 2q13, 5p15, 12p13.33, 14q32, and 22q11 regions, and rare single nucleotide variants (SNVs) in the MYT1, SF3B2, AMIGO2, and ZYG11B genes have been described.5–11
Among the genomic imbalances described in CFM, the 22q11 region has been the most commonly altered in individuals with this diagnosis.5,12–19 The 22q11 region is liable to nonallelic homologous recombination (NAHR) because of the presence of low copy repeats (LCR) named from A to H. 20 Rearrangements proximal to LCR-A, resulting in duplication or triplication at the 22q11.21 region, are the cause of Cat-Eye syndrome (CES) (OMIM # 115470), usually due to a supernumerary marker chromosome (SMC) that contains two extra copies of the chromosome 22q11.1q11.21 region. More rarely, intrachromosomal duplication or triplication of this region can also give rise to a similar phenotype. 21
CES and CFM share many clinical features, such as preauricular tags, stenosis of the external auditory canal, facial asymmetry, mandibular and maxillary hypoplasia, congenital heart disease, and urogenital abnormalities.18,22 In this study we describe three patients with CES, two of whom were previously diagnosed with CFM.
Case Reports
Subject 01
The proband was a girl, the third child of healthy and nonconsanguineous parents, with no significant family history. Her parents were forty years old at conception, and her mother presented with an hypertensive disorder during pregnancy. The patient was delivered at term, presented with laryngomalacia and prolonged neonatal jaundice. She developed respiratory distress and deglutition disorder, needing a percutaneous gastrostomy tube for enteral nutrition. At birth, her weight was 3115 g (−1 SD), length 49 cm (0 SD), and head circumference 34 cm (0 SD). At the first clinical genetic evaluation, at five months and eight days old, her weight was 5140 g (−3 SD), length 61 cm (−2 SD), and head circumference 38 cm (−3 SD). She presented with trigonocephaly (HP:0000243), facial asymmetry (HP:0000324), bilateral microtia (HP:0008551), auditory canal atresia (HP:0000413), preauricular tags (HP:0000384) and pits (HP:0004467), epicanthus (HP:0000286), strabismus (HP:0000486), flat nasal bridge (HP:0005280), anteverted nares (HP:0000463), cleft soft palate (HP:0000185), long philtrum (HP:0000343), micrognathia (HP:0000347) and short neck (HP:0000470) (Figure 1A).

Pictures of the face of subjects 01 (1A), 02 (1B), and 03 (1C). Note preauricular pits in all subjects, and preauricular tags in subjects 01 and 02. Subjects 01 and 02 also presented with microtia and mandibular hypoplasia.
Complementary exams were performed: computed tomography of mastoids revealed bilateral atresia of the auditory canal (HP:0000413), hypoplasia of the mandibular condyle and coronoid process on the right (HP:3000078); echocardiography and electrocardiograph showed interventricular communication (HP:0001629), interatrial communication (HP:0001631), significant pulmonary valve stenosis (HP:0001642), mild aortic valve stenosis (HP:0001646) patent ductus arteriosus (HP:0001643), ascending aortic aneurism, and total atrioventricular block (HP:0001678); whole-spine radiograph was normal; ophthalmologic evaluation showed iris (HP:0000612), macular (HP: HP:0001116) and optic disc (HP: HP:0000588) coloboma of the left eye and abnormality of ocular abduction (HP:0011347); abdominal ultrasonography showed multicystic kidneys and right-sided dysplasia (HP:0000003). She had a clinical diagnosis of Craniofacial Microsomia.
The G banding karyotype of peripheral blood revealed 47,XX,+mar[20]. The Multiplex Ligation-dependent Probe Amplification (MLPA) method, using the P250-B2 kit (MRC-Holland®, Amsterdam, The Netherlands), revealed a triplication of the IL17RA, SLC25A18, BID, MICAL3, and USP18 probes, all mapped at 22q11.2, within the CES region. The Chromosomal microarray analysis (CMA), performed with the CytoScan 750 K Array platform (Affymetrix®, Santa Clara, CA, USA) showed a triplication at 22q11.2q11.21 of 1,7 Mb – arr [GRCh37]22q11.1q11.21(16,888,899_18,649,190)x4 – and a duplication at 2p12 of 6,0 Mb – arr[GRCh37]2p12(75,899,977_81,916,909)x3 (Figure 2A). Due to complications of congenital heart disease, the patient died at one year of age.
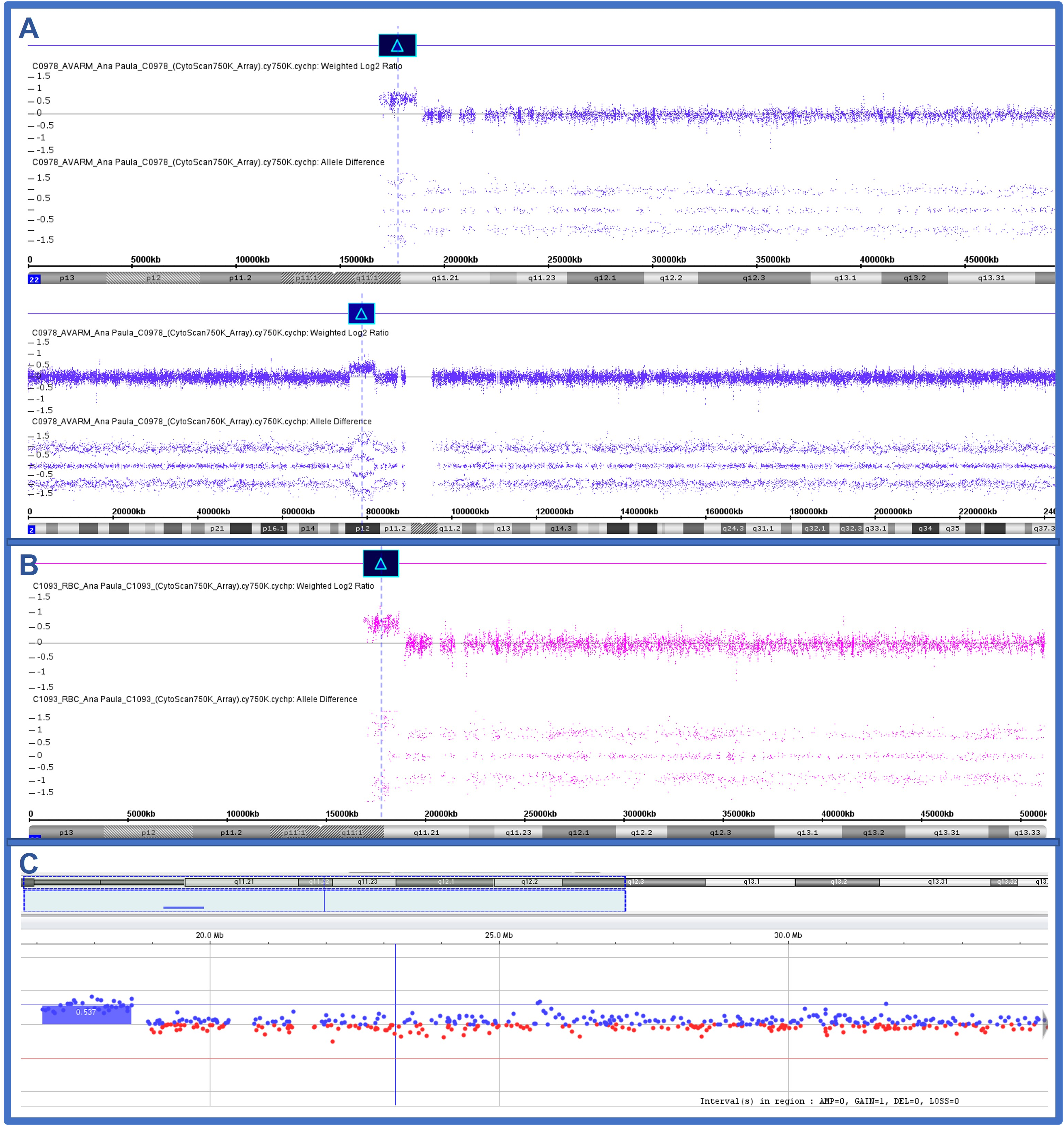
Chromosomal microarray results of subject 01 (2A) showing the tetrasomy at 22q11.2q11.21 (1,7 Mb) and a duplication at 2p12 (6,0 Mb); subject 02 (2B) the tetrasomy at 22q11.2q11.21 (1,7 Mb); and subject 03 (2C) the mosaic of the tetrasomy at 22q11.2q11.21.
Subject 02
The proband is a girl, the second child of healthy and nonconsanguineous parents, with no significant family history. Her mother was forty-three years old, and her father was forty-seven. The pregnancy was uneventful, and there were no known exposures to tobacco and alcohol. Her mother was medicated with clonazepam and topiramate at the beginning of the gestational period (until six weeks). She was delivered by cesarean section at 34 weeks, and her weight was 1780 g (–0.99 SD), length 41.5 cm (–1.7 SD), head circumference 31 cm (–0.05 SD), and Apgar score 8/8. At birth, a right ear malformation was observed. She was hospitalized for 34 days with O2 support. Her echocardiogram was normal. She was breastfeeding until five months. The patient presented with psychomotor developmental delay. She held her head steady unsupported at six months of age, sat without support at eight months, and walked at twelve months. She received physiotherapy twice a week.
At six months of age, the physical evaluation revealed weight 5570 g (–2.5 SD), length 61 cm (–2.74 SD) and head circumference 42 cm (−1.86 SD), microtia first degree on the right (HP:0011266), preauricular pit on the right (HP:0004467), preauricular tags on the left (HP:0000384), narrow external auditory canal (HP:0000402), epicanthus inversus (HP:0000537), mandibular asymmetry, abnormality on the oral commissure on the left, umbilical hernia (HP:0001537) and sacral dimple (HP:0000960) (Figure 1B). Complementary exams were performed: computed tomography of the face revealed hypoplasia in the mandibular coronoid process, parotid gland, and temporal muscle to the right. Deformities were also observed in the right auricular pavilion, and the external auditory canal was obliterated due to apparent atresia. The X-ray of the total column showed T7 and T10 hemivertebrae with secondary scoliosis on the left, probably a malformation in the cervicothoracic transition. The ultrasound of the total abdomen was normal, and the ophthalmologic evaluation was normal. She had a clinical diagnosis of craniofacial microsomia.
The G banding karyotype of peripheral blood revealed 47,XX,+idic(22)(q11.2)[20]. The MLPA method, using the P250-B2 kit (MRC-Holland®, Amsterdam, The Netherlands) revealed a triplication of the IL17RA, SLC25A18, BID, MICAL3, and USP18 probes, all mapped at 22q11.2, within the CES region. The CMA, performed with the CytoScan 750 K Array platform (Affymetrix®, Santa Clara, CA, USA) confirmed a triplication at 22q11.1q11.21 of 1,7 Mb – arr GRCh37]22q11.1q11.21(16,888,899_18,649,190)x4 (Figure 2B).
Subject 03
The proband is a boy, the first child of healthy and non-consanguineous parents. His mother was 32 years old, and his father was 45. The pregnancy was uneventful, and he was delivered by cesarean section. At birth, his weight was 3045 g (–0.64 SD), length 50 cm (0.06 SD), head circumference 36 cm (1.21 SD), and Apgar 8/9. The patient had corrective surgery for an unperforated anus and was discharged after ten days. At fifteen days, he returned to the intensive care unit with a genitourinary infection, and at approximately forty days, he was diagnosed with biliary atresia that progressed to chronic hepatopathy. After fifty-six days, he underwent Kasai portoenterostomy. At physical evaluation, at two years and five months, his weight was 13.2 kg (0.14 SD), length 93 cm (0.71 SD), and head circumference 53,5 cm (3.43 SD); he presented with plagiocephaly (HP:0001357), prominent forehead (HP:0011220), preauricular pits on the right (HP:0004467), long eyelashes (HP:0000527), divergent strabismus (HP:0020049), single transverse palmar crease (HP:0000954), umbilical hernia (HP:0001537), and cryptorchidism (HP:0000028) (Figure 1C). He also presented with psychomotor developmental delay (HP:0001263).
Echocardiography revealed persistent superior vena cava (HP:0005301), ostium secundum (HP:0001684), atrial septal defect (HP:0001631), and pulmonary hypertension. The G banding karyotype of peripheral blood revealed 47,XY,+mar[29]/46,XY[21]. His mother and father presented normal karyotype. The CMA, performed with the Agilent SurePrint G3 Human CGH Microarray 8 × 60 K (Agilent Technologies, Santa Clara, CA, USA), revealed a duplication at 22q11.1q11.2 of 1.5 Mb – arr[GRCh37] 22q11.1q11.2(17,096,855–18,641,468)x3 (Figure 2C). However, since he presents chromosomal mosaicism, he probably presents a triplication of the 22q11.1q11.2 region, which was masked in the CMA experiment due to the presence of normal cells.
Discussion
Individuals with CES and clinical diagnosis of CFM have been previously described and present with preauricular tags or pits and facial asymmetry/hemifacial microsomia as the most common features.18,23,24 In addition, patients with CES frequently present anorectal and urogenital malformations, ocular coloboma, and congenital heart disease. On the other hand, microtia and hemifacial microsomia and/or facial asymmetry have been described as the most frequent phenotypes in CFM. 25 However, in both conditions (CES and CFM), these most common features are not necessarily expressed in combination. Although there are studies of individuals with CFM presenting 22q11 rearrangements and individuals with CES presenting CFM features, this association is probably underestimated. 21
In this study, subjects 01 and 02, primarily referred due to a clinical diagnosis of CFM, presented with microtia, preauricular tags and pits, and mandibular hypoplasia. Subject 03, who did not have a clinical diagnosis of CFM, only had preauricular pits. CMA revealed a similar triplication of approximately 1.7 Mb at 22q11.1q11.21, corresponding to the CES region, in all of them. In the literature review, it is possible to observe that preauricular tags or pits were reported in all the cases with CES and CFM (Table 1). Besides, subjects 01 and 02 of the present study also presented with microtia, and hearing loss was found in subject 01 and in two other patients previously reported. Ocular coloboma, a frequent sign in CES patients, was found only in subject 01. Although the name of this condition was defined based on the presence of iris coloboma, it may be absent in almost half of the individuals with CES. 26 Subjects 01 and 02 and two other reported patients (Table 1) presented with mandibular hypoplasia. Therefore, the most frequent craniofacial features found in individuals with CFM and CES are preauricular tags and/or pits, which is also the most common feature in CES, and mandibular hypoplasia (Table 1). However, since the clinical phenotype is heterogeneous, genetic tests should be performed for individuals with a suspicion of CES, even in those no presenting with the classical clinical features of the syndrome, such as iris coloboma. In addition, many individuals without a clinical hypothesis of CES can also be identified after the diagnostic investigation using genomic approaches, such as CMA.
Comparison of Clinical Features Between the Patients of This Study and Other CFM/OAVS Patients with CES Described in the Literature.
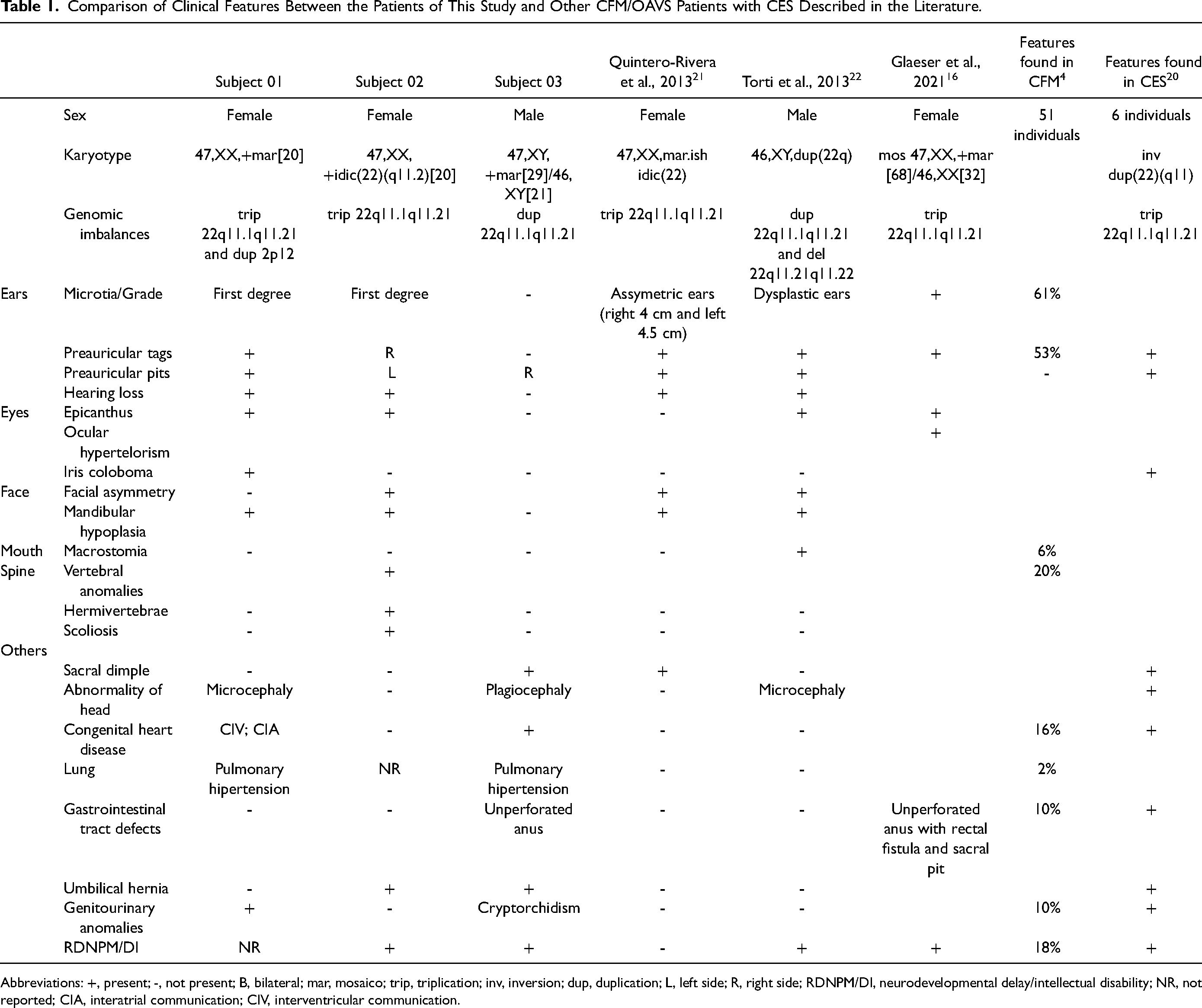
Abbreviations: +, present; -, not present; B, bilateral; mar, mosaico; trip, triplication; inv, inversion; dup, duplication; L, left side; R, right side; RDNPM/DI, neurodevelopmental delay/intellectual disability; NR, not reported; CIA, interatrial communication; CIV, interventricular communication.
Other features, outside the craniofacial region, included vertebral anomalies, found only in subject 02; microcephaly and plagiocephaly; congenital heart disease, found in subjects 01 and 03; unperforated anus, found in subject 03 and another reported patient, which is a well-known feature in CES; umbilical hernia, found in subjects 02 and 03; and genitourinary anomalies, found in subjects 01 and 03. Among these features, it is worthy to mention that congenital heart anomalies, including atrioventricular septal defects and total anomalous pulmonary venous return, are found in about 60% of CES patients. 26 In addition, neurodevelopmental delay was found in subjects 01 and 03 and two other reported patients with CFM and CES (Table 1).
Subject 01 had another CNV, a rare duplication of 6.0 Mb at 2p12 including 27 genes, classified as a variant of unknown clinical significance (VUS). Although no other individuals with this specific duplication were found in the literature, for a genotype-phenotype correlation, this individual presented with a more severe phenotype and deceased before completing one year of age due to complications of congenital heart disease. Considering that all major clinical features presented have already been reported in CES, it is not clear whether the duplication at 2p12 contributed or not to the patient's phenotype.
The CES region contains 28 genes, and 11 of these are included in the OMIM database (XKR3, IL17RA, ADA2, CECR2, SLC25A18, ATP6V1E1, BID, MICAL3, MIR648, PEX26, TUBA8, USP18). Nevertheless, only five of them are associated with a specific phenotype. Knijnenburg et al., 2012, described a three-generation family with CES, in which the index case presented a 600 kb intrachromosomal triplication involving six genes. The authors suggested that the tetrasomy of the CECR2, SLC25A18, and ATP6V1E1 genes, in the 22q11 region, were responsible for anorectal, renal, and preauricular anomalies in the CES. 21 All these genes are encompassed in the triplicated region of the patients herein described, supporting a role for these genes in the CFM phenotype.
In the DECIPHER database, there is a reported patient (289462) with a duplication of 214.64 kb, paternally inherited, presenting transposition of the great arteries and preauricular tag, but no clinical information of the father was available. This duplication encompasses the entire CERC3 gene and the 3’ region of the CERC1/ADA2 (OMIM* 607575
In conclusion, this study corroborates the phenotypic overlapping between CES and CFM/OAVS and strengthens the role of the 22q11 genomic region in these phenotypes. We reinforce that the candidate genes for CFM/OAVS features, particularly ear malformation, preauricular tags/pits, and facial asymmetry, can be located in the proximal region of the 22q11.2 region.
Footnotes
Acknowledgments
The authors thank the patients and their families.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Statement
This study was approved by the local Ethics Committee Board. Written informed consent and permission to use the images were obtained from the parents.
Funding
The study was supported by the Conselho Nacional de DesenvolvimentoCientífico e Tecnológico – CNPq (#460422/2014-6), Fundo de Apoio ao Ensino,Pesquisa e Extensão/UNICAMP (#519.292–0144/15; # 519.294–0411/15), and Fundação de Amparo à Pesquisa do Estado de Alagoas – FAPEAL (#60030000856/2016). SSS was granted by Coordenação de Aperfeiçoamentode Pessoal de Nível Superior – CAPES (#01-P-3368/2017), VLGSL and TMF are researchers granted by CNPq (#309782/2020-1 and #306861/2019-4, respectively).
Supplemental Material
Supplemental material for this article is available online.