
Abstract
TFE3-rearranged perivascular epithelioid cell tumors (PEComas) of the kidney are rare, with fewer than 50 examples reported in the literature. A 32-year-old man presented with an 8.2 cm right renal mass and bulky retroperitoneal lymphadenopathy, raising concern for lymphoma versus renal cell carcinoma (RCC). An initial renal biopsy performed at an outside institution was diagnosed as “RCC, favor chromophobe type,” based on morphological features and KIT positivity. Due to limited tumor cells in the initial biopsy for additional studies, a repeat renal biopsy and a biopsy of a right retroperitoneal lymph node were obtained at our institution. Both specimens revealed nests and sheets of epithelioid tumor cells with abundant granular eosinophilic to clear cytoplasm and a perivascular growth pattern. Focal areas showed prominent cell membranes, and melanin pigment was identified in rare tumor cells. Immunohistochemical staining demonstrated strong, diffuse nuclear positivity for TFE3, as well as positivity for HMB45, cathepsin K, and KIT. The tumor cells were negative for pan-keratin, keratin 7, PAX8, CA9, CD10, and SMA. A next-generation sequencing-based fusion assay identified an SFPQ::TFE3 gene fusion. The tumor was classified as a TFE3-rearranged PEComa. The patient underwent systemic therapy followed by radical nephrectomy. The tumor invaded the renal sinus fat and showed extensive lymphovascular invasion (ypT3aN1). Despite treatment, restaging imaging at the 16-month follow-up revealed liver metastases. Subsequent imaging demonstrated further progression, and the patient ultimately opted for supportive care at 32 months. This report highlights the diagnostic challenges of renal TFE3-rearranged PEComas, due to their rarity and overlapping morphologic, immunophenotypic, and molecular features with other neoplasms.
Introduction
Perivascular epithelioid cell tumors (PEComas) are a family of mesenchymal neoplasms characterized by perivascular epithelioid cell differentiation. This family includes angiomyolipoma (AML), lymphangioleiomyomatosis (LAM), PEComas of the lung (including those formerly known as clear cell sugar tumors), as well as PEComas arising in other visceral sites, particularly the gastrointestinal tract and uterus, and in soft tissue.1–4 These tumors are more common in women, with peak incidence occurring in young to middle-aged adults. 4 Histologically, they are typically composed of nests and sheets of epithelioid (occasionally spindled) cells with clear to eosinophilic cytoplasm, often demonstrating close association with blood vessels. 3
PEComas characteristically express melanocytic markers, including HMB45 (the most sensitive immunomarker), melan-A, and MiTF, as well as muscle markers such as smooth muscle actin (SMA) and desmin. Cathepsin K staining is typically strong and diffuse. 5 There is a strong association between certain members of the PEComa family, specifically AML and LAM, and tuberous sclerosis complex (TSC). A high frequency of both syndromic and sporadic PEComas have shown inactivation of the TSC1 or TSC2 genes, leading to activation of the mammalian target of rapamycin (mTOR) pathway.6,7 Additionally, a small subset of PEComas harbor TFE3 gene fusions.1,8 To date, fewer than 100 TFE3-rearranged PEComas have been reported in the English-language literature, with over 30 arising in the kidney, making it the most frequently affected site.8,9
We present a TFE3-rearranged PEComa of the kidney and discuss the associated diagnostic challenges.
Case Report
A 32-year-old previously healthy man presented with gross hematuria and flank pain. Imaging revealed an 8.2 cm right renal mass with bulky retroperitoneal adenopathy, raising concern for lymphoma versus renal cell carcinoma (RCC).
An initial renal biopsy performed at an outside institution was diagnosed as “RCC, favor chromophobe type.” Upon review of the outside slides, the biopsy showed nests of epithelioid tumor cells with abundant granular eosinophilic to clear cytoplasm, focally prominent cell membranes, and round to oval hyperchromatic nuclei (Figure 1A and B). Immunohistochemical (IHC) staining performed at the outside institution demonstrated that the tumor cells were diffusely positive for KIT (Figure 1C) and AMACR (Figure 1D), and negative for pan-keratin, keratin 7, PAX8, carbonic anhydrase 9 (CA9), and DOG1.
An initial renal biopsy performed at an outside institution showed nests of epithelioid tumor cells with abundant granular eosinophilic to clear cytoplasm and round to oval hyperchromatic nuclei (A and B). Focal distinct cell borders were present (A, arrows). Immunohistochemical staining performed at the outside institution demonstrated that the tumor cells were diffusely positive for KIT (C) and AMACR (D).
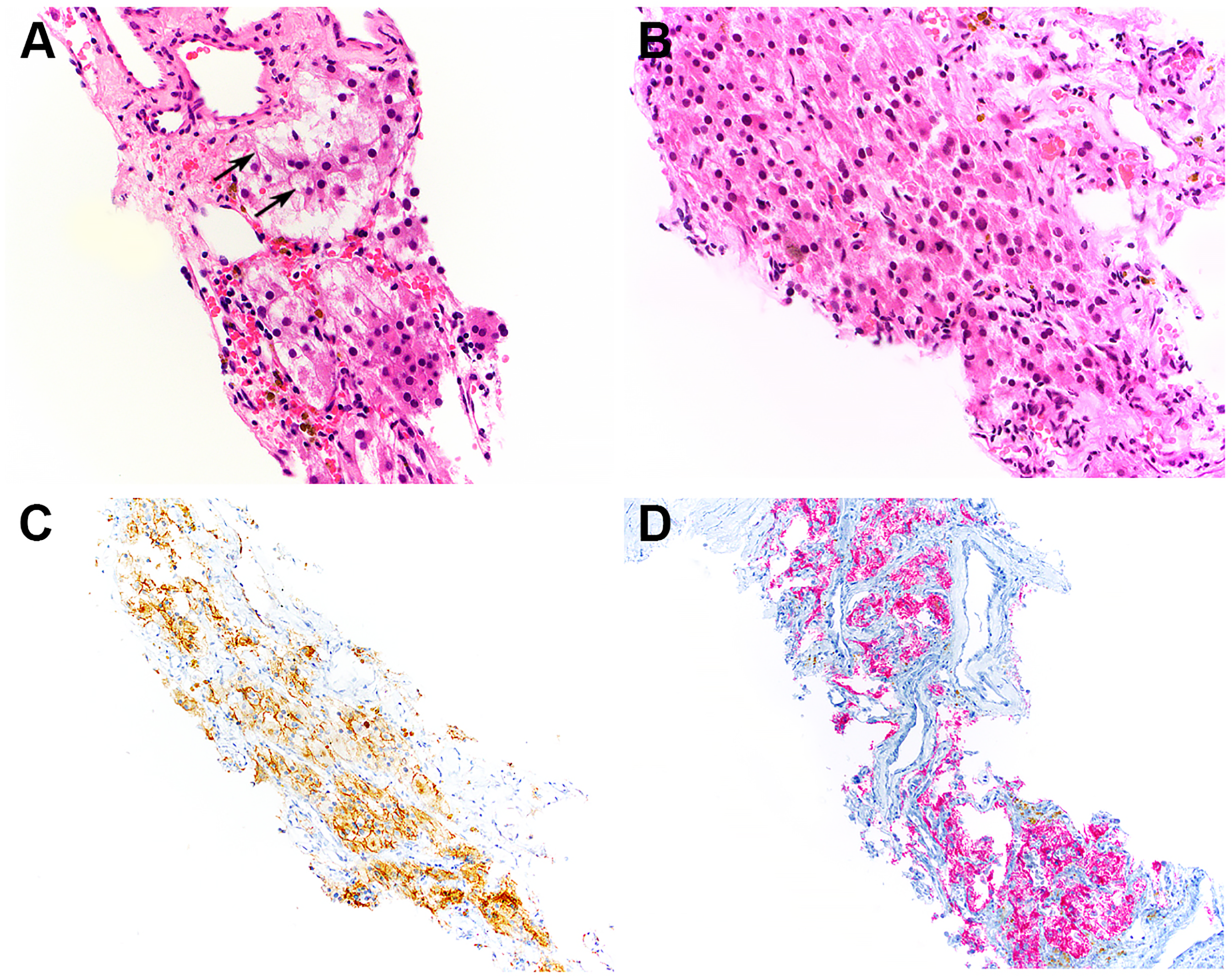
Due to limited tumor cells in the initial biopsy for additional studies when the patient came to our institution for treatment, a repeat renal biopsy and a biopsy of a right retroperitoneal lymph node were performed. Both specimens showed nests and sheets of epithelioid tumor cells with abundant granular eosinophilic to clear cytoplasm, exhibiting a perivascular growth pattern (Figure 2A-D). Focal areas of the tumor showed cells with abundant clear to eosinophilic cytoplasm and well-defined cell borders (Figure 2B). Melanin pigment was identified in rare tumor cells (Figure 2C). The majority of tumor cells exhibited monotonous, hyperchromatic, round to oval nuclei, although occasional cells demonstrated nuclear enlargement with irregular contours and prominent nucleoli (Figure 2D). IHC staining demonstrated strong, diffuse nuclear positivity for TFE3 (Figure 2E), as well as positivity for HMB45 (Figure 2F), cathepsin K (Figure 2G), and KIT (Figure 2H; predominantly membranous). The tumor cells were negative for pan-keratin AE1.3/CAM5.2, PAX8 (Figure 2F inset), CA9, CD10, keratin 7, MiTF, Melan-A, MART-1, SMA, muscle-specific actin, SF1, and inhibin. Fontana-Masson stain highlighted intracellular melanin pigment (Figure 2C inset). Next-generation sequencing-based fusion assay identified an SFPQ::TFE3 gene fusion, involving SFPQ Exon 9 and TFE3 Exon 5.

Repeat biopsies showed nests and sheets of epithelioid tumor cells with abundant granular eosinophilic to clear cytoplasm, arranged in a perivascular growth pattern (A). Some tumor cells demonstrated abundant clear to eosinophilic cytoplasm and prominent cell membranes (B, arrows). Melanin pigment was identified in rare tumor cells (C; inset: Fontana-Masson stain). Occasional tumor cells exhibited nuclear enlargement with irregular contours and prominent nucleoli (D, arrow). Immunohistochemical staining demonstrated strong, diffuse nuclear positivity for TFE3 (E), as well as positivity for HMB45 (F), cathepsin K (G), and KIT (H, predominantly membranous; inset: higher magnification). The tumor cells were negative for PAX8 (F, inset).
Integrating the morphologic and IHC findings from both the outside and in-house biopsies, along with the molecular results, the tumor was classified as a TFE3-rearranged PEComa. The patient was subsequently treated with lenvatinib (a tyrosine kinase inhibitor), everolimus (an mTOR inhibitor), and pembrolizumab (a PD-1 receptor inhibitor). Imaging studies at 4 months following the diagnosis of TFE3-rearranged PEComa showed slight improvement; however, the tumor remained unresectable. Restaging imaging at 6 months demonstrated stable disease, with no further tumor regression or progression. The patient subsequently underwent radical nephrectomy and retroperitoneal lymph node dissection.
Gross examination of the nephrectomy specimen revealed a 12 cm encapsulated mass within the mid to lower pole of the right kidney, abutting the renal pelvis and sinus fat. Sectioning revealed a lobulated, predominantly red-brown to pink, fleshy cut surface with a centrally located yellow-white necrotic area (Figure 3A). Microscopic examination of the nephrectomy specimen revealed a tumor predominantly composed of sheets and nests of epithelioid cells arranged around medium-sized blood vessels and hyalinized septa (Figure 3B). Prominent alveolar growth pattern was identified focally, exhibiting a thin-walled capillary network surrounding nests of clear to slightly eosinophilic tumor cells, mimicking clear cell renal cell carcinoma (CCRCC) (Figure 3C). Focal areas showed variably sized tumor nests with surrounding cleft-like spaces (Figure 3D). In addition, prominent cell membranes, hyperchromatic nuclei, and occasional binucleation were observed, mimicking chromophobe renal cell carcinoma (ChRCC) (Figure 3E). Rare pleomorphic tumor cells with irregular nuclear contours and abundant dense eosinophilic cytoplasm, along with occasional multinucleated tumor cells, were also observed (Figure 3F). Scattered tumor cells contained melanin pigment (Figure 3B and G). Approximately 10% of the tumor showed necrosis. Mitotic activity was rare or absent. IHC staining demonstrated that the tumor cells were diffusely positive for cathepsin K and negative for PAX8. The tumor invaded the renal sinus fat (Figure 3H) and exhibited extensive lymphovascular invasion. Metastatic tumor was present in the hilar (2/2), interaortocaval (6/11), and para-aortic (2/5) lymph nodes. Additionally, a 12 cm soft tissue mass was identified in the interaortocaval region, clinically interpreted as an enlarged interaortocaval lymph node, which upon sectioning demonstrated a vaguely lobulated, red-brown, fleshy cut surface with central to peripheral yellow, soft necrotic areas comprising approximately 35% to 40% of the tumor (Figure 4A). Histologically, this mass predominantly exhibited an alveolar growth pattern (Figure 4B), with tumor nests surrounded by cleft-like spaces focally (Figure 4C). Melanin pigment was present in scattered tumor cells (Figure 4B and D). No lymphoid tissue was identified within the tumor deposit. The tumor was staged as ypT3aN1.

Gross examination of the nephrectomy specimen revealed a 12 cm encapsulated mass within the mid to lower pole of the right kidney. Sectioning revealed a lobulated, predominantly red-brown to pink, fleshy cut surface with a centrally located yellow-white necrotic area (A). The tumor was predominantly composed of sheets and nests of epithelioid cells arranged around medium-sized blood vessels and hyalinized septa (B). Prominent alveolar growth pattern was identified focally, exhibiting a thin-walled capillary network surrounding nests of clear to slightly eosinophilic tumor cells, mimicking clear cell renal cell carcinoma (C). Focal areas showed tumor nests with surrounding cleft-like spaces (D). Prominent cell membranes (arrowheads), hyperchromatic nuclei, and occasional binucleation (arrow) were observed, mimicking chromophobe renal cell carcinoma (E). Rare pleomorphic (arrows) and multinucleated (inset, arrow) tumor cells were also noted (F). Scattered tumor cells contained melanin pigment (B, G). The tumor invaded the renal sinus fat (H, arrows).

A 12 cm tumor deposit was identified in the interaortocaval region. Sectioning demonstrated a vaguely lobulated, red-brown, fleshy cut surface with central to peripheral yellow, soft necrotic areas (A). Histologically, this mass predominantly exhibited an alveolar growth pattern (B), with tumor nests surrounded by cleft-like spaces focally (C). Melanin pigment was present in scattered tumor cells (B, D).
Restaging scans performed at the 16-month follow-up revealed new liver metastases, despite continued treatment with lenvatinib and pembrolizumab. These therapies were subsequently discontinued, and the patient was treated with additional immune checkpoint inhibitors, including ipilimumab (a CTLA-4 inhibitor) and nivolumab (a PD-1 receptor inhibitor) for 3 cycles, and subsequently with cabozantinib, a multi-kinase inhibitor targeting VEGFR2, c-MET, and AXL. However, follow-up imaging continued to demonstrate persistent disease with areas of progression. The patient was then enrolled in a Phase 1 clinical trial of NPX267, a humanized monoclonal antibody targeting KIR3DL3, which is involved in co-inhibition of CD8+ T cells and NK cells. This was followed by treatment with sunitinib, a tyrosine kinase inhibitor. Both agents were discontinued due to rising liver function tests. Restaging imaging performed at the 32-month follow-up showed further disease progression. Although not ready to transition to hospice care, the patient opted for supportive management and alternative care.
Discussion
In the kidney, the PEComa family includes AML, and, more rarely, LAM of the renal sinus and intraglomerular lesions.5,9 Classic AML is a benign mesenchymal tumor composed of varying proportions of adipose tissue, spindle and epithelioid smooth muscle cells, and thick-walled blood vessels. Epithelioid AML is a rare variant characterized by the presence of at least 80% epithelioid cells. 10 These tumors typically display sheets or nests of large polygonal epithelioid cells with abundant eosinophilic to clear cytoplasm, often accompanied by prominent nucleoli and occasional multinucleated or pleomorphic forms. Epithelioid AML is considered synonymous with PEComa by some authors,10,11 and the terms are often used interchangeably in the literature.2,3,12–16 The present renal PEComa may represent an epithelioid AML with TFE3 rearrangement. However, not all authors agree that epithelioid AML is synonymous with PEComa, as some tumors exhibit both epithelioid and classic AML components. Classifying such tumors as PEComas may be inappropriate, even though they all fall within the PEComa family. Pure epithelioid AML may more accurately represent PEComas. Further studies are needed to clarify the nomenclature and classification of these entities. Additionally, LAM of the renal sinus is a rare lesion characterized by a plaque-like proliferation of spindle and epithelioid smooth muscle cells, with branching, slit-like vascular channels. 5 Another rare entity, intraglomerular lesions, consists of minute nodules within the glomerular capillary tuft composed of either adipocytes and epithelioid smooth muscle cells or pure epithelioid smooth muscle cells. 5
To date, fewer than 100 TFE3-rearranged PEComas have been reported in the English-language literature, with over 30 arising in the kidney, making it the most frequently affected site.8,9 SFPQ is the most common fusion partner in TFE3-rearranged PEComas. 8 Other fusion partners, including NONO, ASPSCR1, and DVL2, have also been reported. 8 TFE3-rearranged PEComas tend to occur in younger patients, exhibit a prominent alveolar growth pattern with epithelioid morphology, and often lack expression of smooth muscle markers. They do not demonstrate inactivation of TSC1 or TSC2 genes, nor is there a clinical association with tuberous sclerosis.8,17,18 Although data in the literature are limited, TFE3-rearranged PEComas appear to exhibit aggressive behavior. Among 42 reported tumors with clinical follow-up of at least 1 year, 13 (31%) developed distant metastases, 5 (12%) resulted in patient death, and another 5 (12%) showed local recurrence or regional lymph node metastasis. 8
In a recent review of 35 TFE3-rearranged PEComas of the kidney, SFPQ was the most frequent fusion partner (74%), consistent with TFE3-rearranged PEComas overall, followed by ASPSCR1 (16%), RBM10 (5%), and MED15 (5%). 9 A novel RBMX::TFE3 gene fusion, resulting from a paracentric inversion of the X chromosome, was also recently reported in a highly aggressive pediatric renal PEComa. 19 Compared to TFE3-rearranged PEComas at other sites, particularly the uterus, bladder, and soft tissues, renal tumors tend to occur less frequently in women (60% vs 73%), arise in younger patients (second vs fourth decade of life), and less frequently harbor the NONO::TFE3 rearrangement. In contrast, NONO::TFE3 is the second most common alteration after SFPQ::TFE3 in extrarenal TFE3-rearranged PEComas. 9 IHC staining of renal TFE3-rearranged PEComas consistently demonstrates strong expression of cathepsin K (99%) and melanocytic markers, including HMB45 (91%) and MART-1/Melan-A (80%), along with negativity for PAX8, keratins, and S100. Muscle marker expression is limited, with only occasional positivity for SMA (19%) and no reported desmin expression. 9 While the immunoprofile of renal and extrarenal TFE3-rearranged PEComas largely overlaps, it differs slightly from that of PEComas associated with TSC1/TSC2 mutations, which more frequently express muscle markers. Additionally, melanin pigment deposits have been observed in approximately half of renal TFE3-rearranged PEComas, whereas they appear less frequently in their extrarenal counterparts.8,9 The presence of melanin pigment in these tumors has been considered a distinguishing feature from TSC1/TSC2-mutated PEComas, leading some authors to propose reclassifying them as “melanotic Xp11 neoplasms,”20,21 although the term “TFE3-rearranged PEComas” has become more commonly used in recent literature.8,9 Argani et al also proposed the alternative term “TFE3-rearranged PEComa-like neoplasms,” noting that co-expression of melanocytic and muscle markers, a hallmark of conventional TSC-mutated PEComas, is uncommon in TFE3-rearranged tumors, which are also less responsive to mTOR inhibitor therapy. 8 Six of the 14 patients with renal TFE3-rearranged PEComas and available clinical outcome data died of the disease, most after developing metastases. 9
TFE3-rearranged PEComas of the kidney may present significant diagnostic challenges due to their rarity and overlapping morphologic, IHC, and molecular features with RCCs. In the present patient, the focal prominent cell membranes, abundant clear to eosinophilic cytoplasm, hyperchromatic nuclei, and occasional binucleation, along with KIT positivity, initially led to a misdiagnosis of ChRCC. KIT expression has been reported in a small subset of PEComas. In a case series of soft tissue and gynecologic PEComas, KIT positivity was observed in 1 of 20 tumors. 4 Additionally, among 61 previously reported PEComas, 2 of 6 tumors tested for KIT expression were positive. 4 Cathepsin K positivity, as seen in the present tumor, can also be observed in ChRCC, although typically with less intensity. 22 However, the absence of pan-keratin, keratin 7, and PAX8 expression in this tumor argues against ChRCC. The tumor also exhibited an alveolar growth pattern with a capillary network surrounding nests of tumor cells with clear to slightly eosinophilic cytoplasm, mimicking CCRCC. Nonetheless, the lack of expression of pan-keratin, PAX8, CA9, and CD10 helps to exclude CCRCC. Furthermore, although rare PEComas may show focal anomalous keratin expression, 2 they do not show the diffuse keratin expression seen in RCCs. AMACR expression can be seen in several renal tumors, including papillary RCC, mucinous tubular and spindle cell carcinoma, variably in CCRCC, and rarely in oncocytoma and ChRCC. 23 Although the morphology of the present tumor does not resemble papillary RCC or mucinous tubular and spindle cell carcinoma, it contains areas that mimic CCRCC, oncocytoma, and ChRCC. The combination of positive KIT and negative keratin 7 expression also raises suspicion for oncocytoma. To date, AMACR expression has only been evaluated in a retroperitoneal sclerosing PEComa, where it was negative. 24 The present tumor represents the first reported example of a PEComa with positive AMACR expression. Further studies are needed to clarify the expression patterns of AMACR in PEComas, particularly those with TFE3 rearrangements.
Furthermore, the clear to eosinophilic cytoplasm, scattered melanin pigment, diffuse and strong nuclear TFE3 positivity, expression of Cathepsin K and melanocytic marker, and the presence of an SFPQ::TFE3 gene fusion raised suspicion for TFE3-rearranged RCC. SFPQ is one of the 3 most common fusion partners in TFE3-rearranged RCC, along with PRCC and ASPSCR1. TFE3-rearranged RCCs may underexpress epithelial markers such as keratin and EMA. However, they consistently express PAX8, which is negative in TFE3-rearranged PEComas.8,9,25 Although solid nested areas may be present in TFE3-rearranged RCC, papillary architecture is more commonly observed, a feature not seen in TFE3-rearranged PEComa. The pathogenesis of TFE3-rearranged PEComa and RCC may overlap. One hypothesis suggests that if a TFE3 translocation occurs in a renal tubular stem cell, the resulting neoplastic transformation may lead to an RCC expressing PAX8. Conversely, if the same translocation arises in a mesenchymal stem cell, it may give rise to a PEComa. 9 However, in a mouse model, overexpression of the SFPQ::TFE3 gene fusion in renal tubular cells led to the loss of all epithelial markers and a shift toward a PEComa-like phenotype. 26 This finding suggests that some renal TFE3-rearranged PEComas may originate from renal tubular cells, despite their nonepithelial phenotype. Further studies are warranted to elucidate the pathogenesis of renal TFE3-rearranged neoplasms.
TFE3-rearranged PEComas of the kidney should also be differentiated from adrenocortical carcinoma, given the adjacent anatomic location and overlapping morphology, including sheets and nests of tumor cells with eosinophilic to clear cytoplasm. However, negative SF1 and inhibin staining helps to argue against adrenocortical carcinoma, as in the present patient. Clear cell (glycogen-rich) urothelial carcinoma is another important differential diagnosis. It is characterized by an accumulation of glycogen within tumor cells, resulting in a clear cell appearance, and may demonstrate a nested or sheet-like growth pattern. However, negative staining for keratin, p63, and GATA3 supports exclusion of this entity.
Additionally, TFE3-rearranged PEComas of the kidney should be differentiated from alveolar soft part sarcoma (ASPS), given their overlapping morphologic, IHC, and molecular features, although ASPS arising in the kidney is extremely rare. 27 Both TFE3-rearranged PEComas and ASPS can exhibit nested epithelioid tumor cells with eosinophilic granular cytoplasm, surrounded by capillary vasculature forming an alveolar growth pattern. Both can also show diffuse and strong nuclear TFE3 positivity, Cathepsin K expression, and harbor ASPSCR1::TFE3 fusions. 8 However, ASPS typically lacks expression of melanocytic markers. 28 Given the expression of melanocytic markers and overlapping morphology, renal TFE3-rearranged PEComas should also be distinguished from melanoma. In most tumors, melanoma can be separated from PEComas by their strong S100 protein expression and lack of SMA positivity, although TFE3-rearranged PEComas are often negative for smooth muscle markers, as observed in the present tumor. Moreover, clear tumor cells and arborizing fibrovascular septa can be seen in clear cell sarcoma of the kidney, a rare malignant renal tumor of childhood that is molecularly characterized by BCOR alterations, either internal tandem duplication or BCOR::CCNB3 gene fusion,29,30 or by YWHAE::NUTM2 gene fusion.
Like other TFE3-rearranged tumors, TFE3-rearranged PEComas typically show strong TFE3 immunoreactivity by IHC staining; however, discordance between IHC and molecular testing occurs. In a study of PEComas involving both renal and nonrenal sites, 4 nonrenal PEComas demonstrated TFE3 gene rearrangements by fluorescence in situ hybridization (FISH), and all 4 showed strong TFE3 positivity by IHC staining. None of the remaining 24 PEComas (renal and nonrenal) showed TFE3 rearrangements by FISH, although 4 tumors (17%) exhibited moderate to strong TFE3 immunoreactivity. 1 In another study, TFE3 rearrangement was detected by FISH in only 1 of 4 renal PEComas/epithelioid AMLs, despite all showing moderate to strong TFE3 expression by IHC staining. 31 This discordance may be explained by functional upregulation of TFE3 secondary to activation of the mammalian target of rapamycin complex 1 (mTORC1). 31 TRIM63 in situ hybridization (ISH) has also been explored as a screening tool, demonstrating high sensitivity (100%) but low specificity (38%) for TFE3-rearranged PEComa/epithelioid AML. 32
Currently, there are no universally accepted diagnostic criteria for malignant PEComas that are applicable across all organ sites. However, recurrence and/or metastasis has been strongly associated with tumor size greater than 8 cm, mitotic activity exceeding 1/50 high-power fields (HPF), and the presence of necrosis. 4 Folpe et al proposed that PEComas exhibiting 2 or more worrisome features (tumor size >5 cm, infiltrative growth, high nuclear grade and cellularity, mitotic rate ≥1/50 HPF, necrosis, and vascular invasion) can be classified as malignant.2,4 In a series of 16 gynecologic PEComas, Schoolmeester et al suggested modifying the threshold to ≥4 of the following features: gross size ≥5 cm, significant nuclear atypia, necrosis, lymphovascular invasion, and mitotic index ≥1/50 HPF, achieving 100% sensitivity and specificity for identifying malignancy. 33 Bennett et al later proposed lowering the threshold to 3 features in gynecologic PEComas. 34 The PEComa in the present study meets 4 of the 6 criteria proposed by Folpe et al: it measured 12 cm, infiltrated renal sinus fat, and showed necrosis and extensive lymphovascular invasion. However, it falls short of Schoolmeester et al's criteria, with only 3 of 5 features present. Nonetheless, according to Bennett et al's revised threshold, the tumor qualifies as a malignant PEComa. Clinically, this tumor demonstrated aggressive behavior and poor treatment response. Prognostic stratification schemes for renal PEComas/epithelioid AMLs have been explored in several studies. In one study, Brimo et al 35 analyzed 40 renal PEComas/epithelioid AMLs, and proposed a 4-feature predictive model for malignancy: (1) ≥ 70% atypical epithelioid cells (defined as polygonal cells with abundant cytoplasm, vesicular nuclei, nuclear size at least twice that of adjacent nuclei, and prominent nucleoli), (2) ≥ 2 mitotic figures/10 HPF, (3) presence of atypical mitotic figures, and (4) necrosis. The presence of 3 or more of these features was highly predictive of malignant behavior. In another study, Nese et al 36 identified 5 adverse prognostic parameters for renal PEComas/epithelioid AMLs: (1) association with TSC or concurrent classic AML, (2) necrosis, (3) tumor size >7 cm, (4) extrarenal extension and/or renal vein involvement, and (5) a carcinoma-like growth pattern. Based on the number of these features present, tumors were stratified into low risk (<2 features), intermediate risk (2-3 features), and high risk (≥4 features). The PEComa in the current report meets only one of Brimo et al's 4 criteria but satisfies 4 of the 5 adverse prognostic parameters described by Nese et al, suggesting that the latter model may offer more effective risk stratification for renal PEComas. Another question is whether the proposed criteria for PEComas are applicable to TFE3-rearranged PEComas. In a study of 25 TFE3-rearranged PEComas involving multiple organ systems, including the kidney, bladder, and colon, Argani et al identified adverse prognostic factors: size >5 cm, mitotic rate >1/10 HPF, and necrosis. 8 The current tumor meets 2 of these criteria, which may explain its aggressive clinical behavior. Further studies are needed to refine the diagnostic criteria for malignant PEComas.
In conclusion, we present a TFE3-rearranged PEComa of the kidney and highlight the associated diagnostic challenges and potential pitfalls.
Footnotes
Ethical Approval
Ethical approval for this study was granted by the Massachusetts General Hospital (MGH) Institutional Review Board.
Informed Consent
The IRB determined that this research involved minimal risk and approved a waiver for informed consent.
Author Contributions
Ting Zhao: conceptualization, data curation, writing—original draft, writing—review & editing. Kyle M. Devins: conceptualization, data curation, writing—review & editing. Yin P. Hung: writing—review & editing. Chin-Lee Wu: conceptualization, data curation, writing—review & editing.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Trial Registration
Not applicable, because this article does not contain any clinical trials.