
Abstract
Purpose:
Evaluate safety and efficacy of AGN-241622 ophthalmic solution in healthy individuals and individuals with presbyopia.
Methods:
This vehicle-controlled, participant- and investigator-masked, randomized phase 1/2 study enrolled healthy individuals and individuals with presbyopia, aged 40–65 years, at 10 sites in the US from 30 July 2020 to 05 Dec 2022 (NCT04403763). Stage 1 cohorts were randomized (3:1) sequentially to receive AGN-241622 (0.01%, 0.03%, or 0.1% ophthalmical solution) or vehicle once in the left eye. Stage 2a cohorts were randomized (3:1) sequentially to receive AGN-241622 (0.01%, 0.03%, or 0.1%) or vehicle daily in both eyes for 14 days. Safety included treatment-emergent adverse events (TEAEs). Efficacy included change in pupil diameter and high-contrast, binocular distance-corrected near visual acuity.
Results:
A total of 80 participants were randomized to AGN-241622 (n = 61) or vehicle (n = 19), and 76 completed the study; baseline characteristics were well balanced. All TEAEs were of mild severity. All but 1 TEAE (not related to AGN-241622) resolved. In Stage 1, the most frequently reported ocular TEAE was increased lacrimation (AGN-241622 0.01% [n = 4], AGN-241622 0.03% [n = 1], and vehicle [n = 1]). In Stage 2a, there were 2 TEAEs of mild severity (dermatitis) in 1 participant who received AGN-241622 0.1% that led to discontinuation, which resolved and was unrelated to AGN-241622. Modest efficacy in pupil reduction and vision improvement was also observed.
Conclusions:
AGN-241622 ophthalmic solution was well tolerated, and modest efficacy was observed in binocular distance-corrected near visual acuity in presbyopic participants with the drug concentrations assessed in this study.
Introduction
Presbyopia is a common age-related visual system condition that affects an estimated 1.8 billion people in the world and is projected to reach 2.1 billion by 2030.1,2 An irreversible and gradual process that usually begins in individuals aged >40 years, presbyopia is believed to be primarily caused by the loss of accommodative ability of the crystalline lens.3,4 Those with presbyopia may notice that they need to hold reading material further away from their eyes in order to bring the image into focus. Symptoms include double and/or blurred near vision (objects at a distance of 40 cm), ocular discomfort, headache, and eye strain that may lead to fatigue.4–6 Early detection and treatment after onset are critical given that as the condition advances, it may also impair intermediate vision and prevent a range of clear vision. 7
Presbyopia significantly impacts an individual’s quality of life and emotional and mental well-being and has a broader societal impact.8–10 Left uncorrected, individuals may experience a significant effect on activities of daily living that require near and intermediate vision, including reading, writing, smartphone and computer use, and cooking. 9 Current treatment options include eyeglasses, contact lenses, intraocular lens implants, corneal laser procedures, intracorneal implants, scleral alterations, and pharmacological ocular drops;4,5,11,12 however, none of these treatments are fully effective in all patients due to the quality of vision, optical and visual distortions, regression of effect, and other complications.13,14 There remains an unmet need for convenient, reversible, nonsurgical pharmacological treatments that do not compromise visual performance.
One pharmacological approach to treating presbyopia is to induce miosis (pupil constriction). Miotic agents, such as pilocarpine and carbachol, cause the ciliary muscles to contract, which enhances the eye’s depth of focus and improves near vision. 15 These treatments are typically administered as eye drops, providing a noninvasive option for patients. Small aperture or pinhole effects created by miosis can also reduce the impact of presbyopia by increasing the depth of field, thus allowing better vision at varying distances.15,16 In 2021, the US Food and Drug Administration (FDA) approved Vuity (pilocarpine hydrochloride ophthalmic solution 1.25%, Allergan, an AbbVie company) as the first eye drop for the treatment of presbyopia, which increases depth of focus in addition to improving focus on near objects.17,18 Clinical trials investigating other pharmacological treatments using miotic agents or in combination are ongoing. 15
AGN-241622 is a pan-agonist at the α2-adrenergic receptor, which may improve near vision by inhibiting the iris dilator muscle, thereby decreasing pupil size and increasing depth of focus. 19 In humans, ophthalmic α2-adrenergic receptor agonists have been shown to reduce pupil size and are safe and effective treatments used for glaucoma.19,20 Based on these results, AGN-241622 ophthalmic solution (Allergan, an AbbVie company) was investigated for the management of presbyopia. The objective of this study was to evaluate the safety, tolerability, pharmacokinetics, and efficacy of AGN-241622 ophthalmic solution compared with vehicle for the first time in healthy individuals and individuals with presbyopia.
Methods
Study design
The study was a multicenter, randomized, double-masked, vehicle-controlled, phase 1/2 trial conducted in 2 stages at 10 sites in the United States from 30 July 2020 to 05 Dec 2022 (ClinicalTrials.gov, NCT04403763). Healthy participants were sequentially randomized in single ascending dose cohorts in Stage 1. Stage 2a consisted of multiple ascending dose cohorts. This research was reviewed by an independent ethical review board and conforms with the principles and applicable guidelines for the protection of human subjects in biomedical research. All study procedures were conducted in accordance with the Declaration of Helsinki, and all participants provided written informed consent before any study-related procedures were performed.
Study population/eligibility criteria
All participants aged 40–65 years were required to be in good general health. In addition, Stage 2a participants were included if they had a diagnosis of presbyopia and complaints of poor near vision impacting activities of daily living. All eligibility criteria are listed in the Supplementary Data. Key inclusion criteria for Stage 2a included mesopic, high-contrast distance-corrected near visual acuity (DCNVA) of 20/40 (J3) to 20/100 (J10) and photopic, high-contrast, near visual acuity correctable to 20/40 (J3+) or better in each eye at screening. Key exclusion criteria for Stage 2a included any ocular conditions that, in the opinion of the investigator, could affect the safety of the participant or interpretation of results, those who were unwilling to use monofocal spectacles/lenses during the study, and those who had an abnormal pupillary reaction to light.
Randomization
Participant randomization to AGN-241622 ophthalmic solution or vehicle was centrally assigned using an interactive web response system. Stage 1 healthy participants (Cohorts 1–3) were single ascending dose cohorts sequentially randomized (3:1) to receive either AGN-241622 (0.01%, 0.03%, or 0.1%) or vehicle once in the left eye. Stage 2a (Cohorts 4–6) were multiple ascending dose cohorts in which participants were randomized (3:1) sequentially to receive either AGN-241622 (0.01%, 0.03%, or 0.1%) or vehicle once daily in both eyes for 14 days. AGN-241622 and the vehicle were provided in identical bottles so that both participants and study investigators/staff could be masked to the treatment assignment.
Interventions and study visits
Stage 1
Stage 1 participants (Cohorts 1–3) randomized to receive the active intervention received a single drop of AGN-241622 ophthalmic solution (0.01%, 0.03%, or 0.1%) or vehicle in the left eye. Participants were admitted to the study center on the evening before dosing. Randomization was conducted on day 1. In Stage 1, participants received a single dose in the morning of day 1 under direct supervision of study center personnel and after a full day of assessments, stayed overnight and continued further safety assessments on the next day (end of study visit). Upon completion of each cohort, an independent data monitoring committee reviewed unmasked safety and efficacy data to recommend proceeding to the next cohort. The FDA also reviewed study data after completion of each cohort. Prior to proceeding to the next cohort, the sponsor waited for feedback from the FDA.
Stage 2
Stage 2a participants (Cohorts 4–6) were randomized to receive active intervention AGN-241622 (0.01%, 0.03%, or 0.1%) or vehicle once daily in both eyes for 14 days. Randomization was conducted on day 1. For the in-office dosing during (days 1–7, and day 14) participants received all doses under the direct supervision of study center personnel. For home dosing during Stage 2a (days 8–13), compliance was monitored by counting the number of bottles dispensed and returned. Before dispensing the new study intervention, study center personnel made every effort to collect all unused study intervention and bottles. Upon completion of each cohort, an independent data monitoring committee reviewed unmasked safety and efficacy data to recommend proceeding to the next cohort. The FDA also reviewed study data after completion of each cohort. Prior to proceeding to the next cohort, the sponsor waited for feedback from the FDA.
Outcome measures
Safety was assessed by the incidence of adverse events (AEs) and changes in cardiovascular, respiratory, gastrointestinal, and neurological systems, vital signs, 12-lead electrocardiogram, and clinical laboratory assessments. Treatment-emergent adverse events (TEAEs) were defined as events with onset or worsening (increased in severity or became serious) on or after the date of the first dose of study intervention.
In Stage 1, mesopic pupil diameter was measured after administration of AGN-241622 or vehicle once in the left eye in each individual eye in healthy participants. In Stage 2a, efficacy assessments included mesopic pupil diameter (with distance correction) measured in the dominant eye, the proportion of participants gaining ≥3 lines in mesopic, high-contrast, binocular DCNVA, and mean change from baseline in mesopic, high-contrast, binocular DCNVA with AGN-241622 compared with vehicle.
Statistical analysis
The safety population included all participants who received 1 or more administration of study intervention. The number and percentage of participants reporting TEAEs for each cohort were tabulated by system organ class, preferred term, and severity. Descriptive statistics were used to assess other safety endpoints. Efficacy analyses were conducted based on the intent-to-treat (ITT) population.
Sample size
The sample sizes for each cohort were empirically determined and sufficient to assess safety and efficacy (as applicable) of each dose of AGN-241622 ophthalmic solution.
Results
A total of 166 participants were screened, 80 participants were randomized to AGN-241622 (n = 61) or vehicle (n = 19), and 76 completed the study (Fig. 1). Of 92 participants screened in Stage 1, 38 were randomized to AGN-241622 (Cohort 1 [n = 9], Cohort 2 [n = 10], Cohort 3 [n = 9]) or vehicle (Cohort 1 [n = 3], Cohort 2 [n = 4], Cohort 3 [n = 3]). In Stage 2a, 74 participants were screened, and 42 were randomized to either AGN-241622 (Cohort 4 [n = 12], Cohort 5 [n = 11], Cohort 6 [n = 10]) or vehicle (Cohort 4 [n = 3], Cohort 5 [n = 3], Cohort 6 [n = 3]) (Fig. 1). Demographics and baseline characteristics were generally balanced and are reported in Table 1. In Stage 1, 1 participant in Cohort 1 (AGN-241622 0.01%) and 2 participants in Cohort 2 (AGN-241622 0.03% [n = 1], vehicle [n = 1]) groups discontinued treatment after randomization and did not receive treatment; 1 participant in Stage 2a Cohort 6 (AGN-242622 0.1%) discontinued due to an AE (Fig. 1).
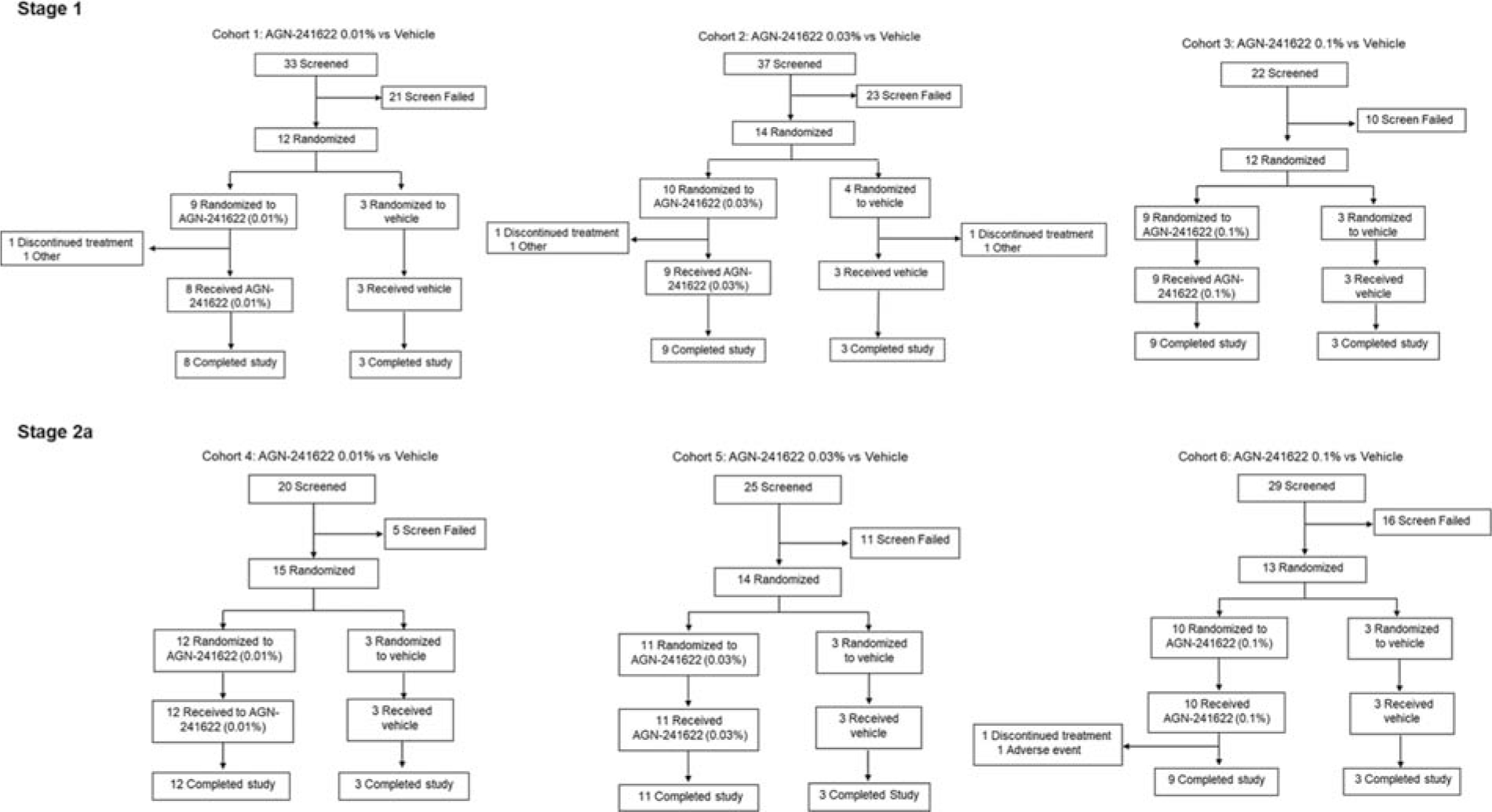
Participant Disposition.
Participant Demographics and Baseline Characteristics (ITT Population)
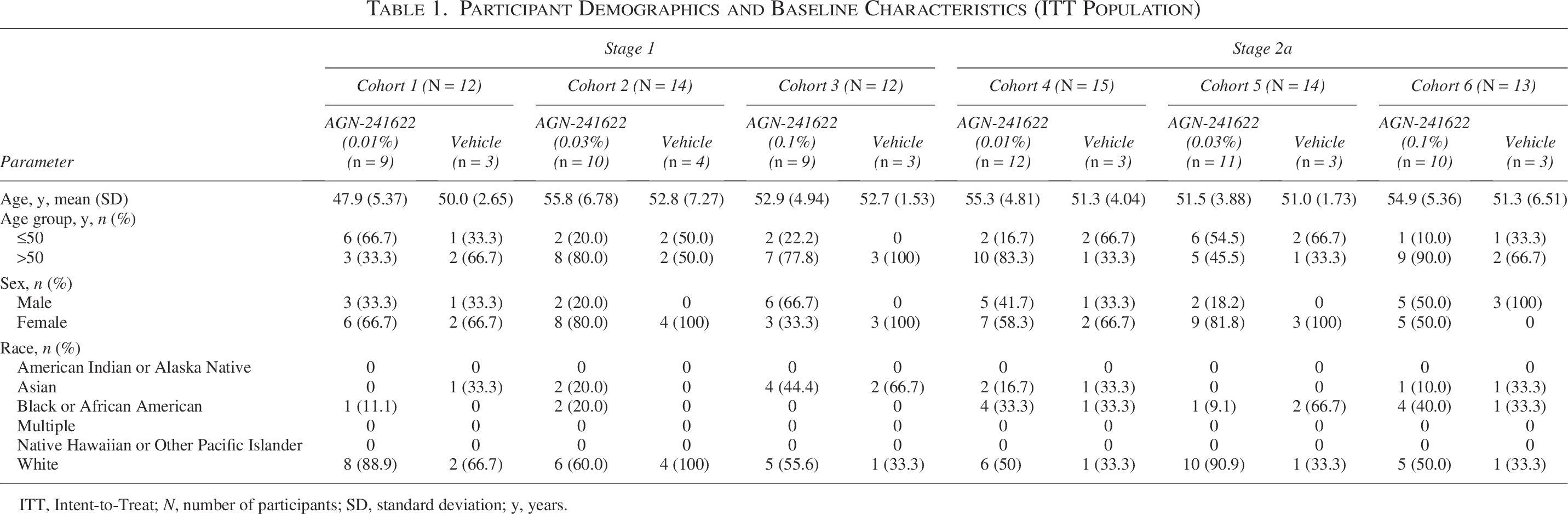
ITT, Intent-to-Treat; N, number of participants; SD, standard deviation; y, years.
In Stage 1, participants in Cohorts 1 (n = 8), 2 (n = 9), and 3 (n = 9) received 1 dose of AGN-241622 0.01%, 0.03%, 0.1%, respectively, in the left eye. Participants in Stage 2a received either AGN-241622 at dose strengths ranging from 0.01% to 0.1% or vehicle once daily in both eyes for 14 days. Cohorts 4 (n = 12), 5 (n = 11), and 6 (n = 10) received AGN-241622 0.01%, AGN-241622 0.03%, or AGN-241622 0.1%, respectively (Table 2). Mean (SD) exposure for participants randomized to AGN-21622 0.01%, AGN-241622 0.03%, and AGN-241622 0.1% in Stage 2a was 14.2 (1.19), 14.7 (0.65), and 14.4 (2.80) days, respectively (Table 2).
Extent of Exposure (Stage 2a Safety Population)
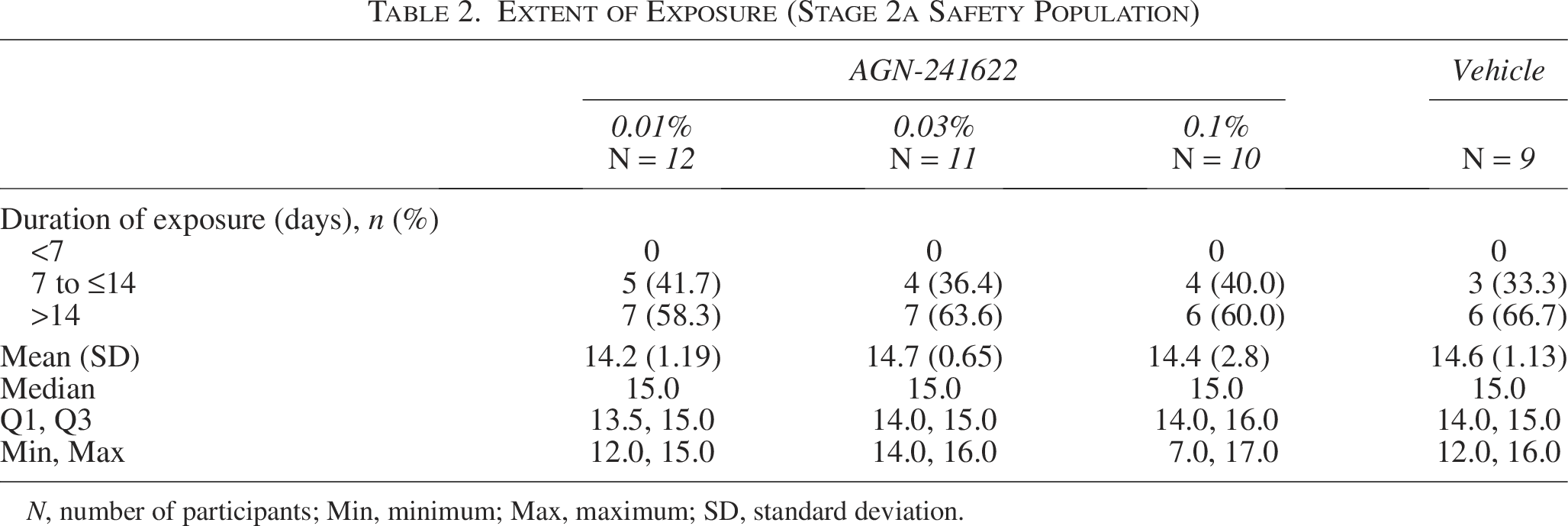
N, number of participants; Min, minimum; Max, maximum; SD, standard deviation.
Efficacy
AGN-241622 resulted in a modest change in pupil diameter assessed under mesopic conditions at post-dose time points in Stage 1, which was consistent across study visits (Fig. 2). Similar results were observed in Stage 2a, in which pupil diameter in each eye was assessed with distance correction under mesopic conditions in the dominant eye and was consistent across all post-baseline timepoints (Fig. 3). At peak efficacy, the decrease from baseline was 23.8% (−1.2 mm from 5.05 mm) in the AGN-241622 0.3% and 20.4% (−1.03 mm from 5.05 mm) in the AGN-241633 0.1% cohorts. In addition, a modest dose response in reduction of pupil diameter size between cohorts was observed.
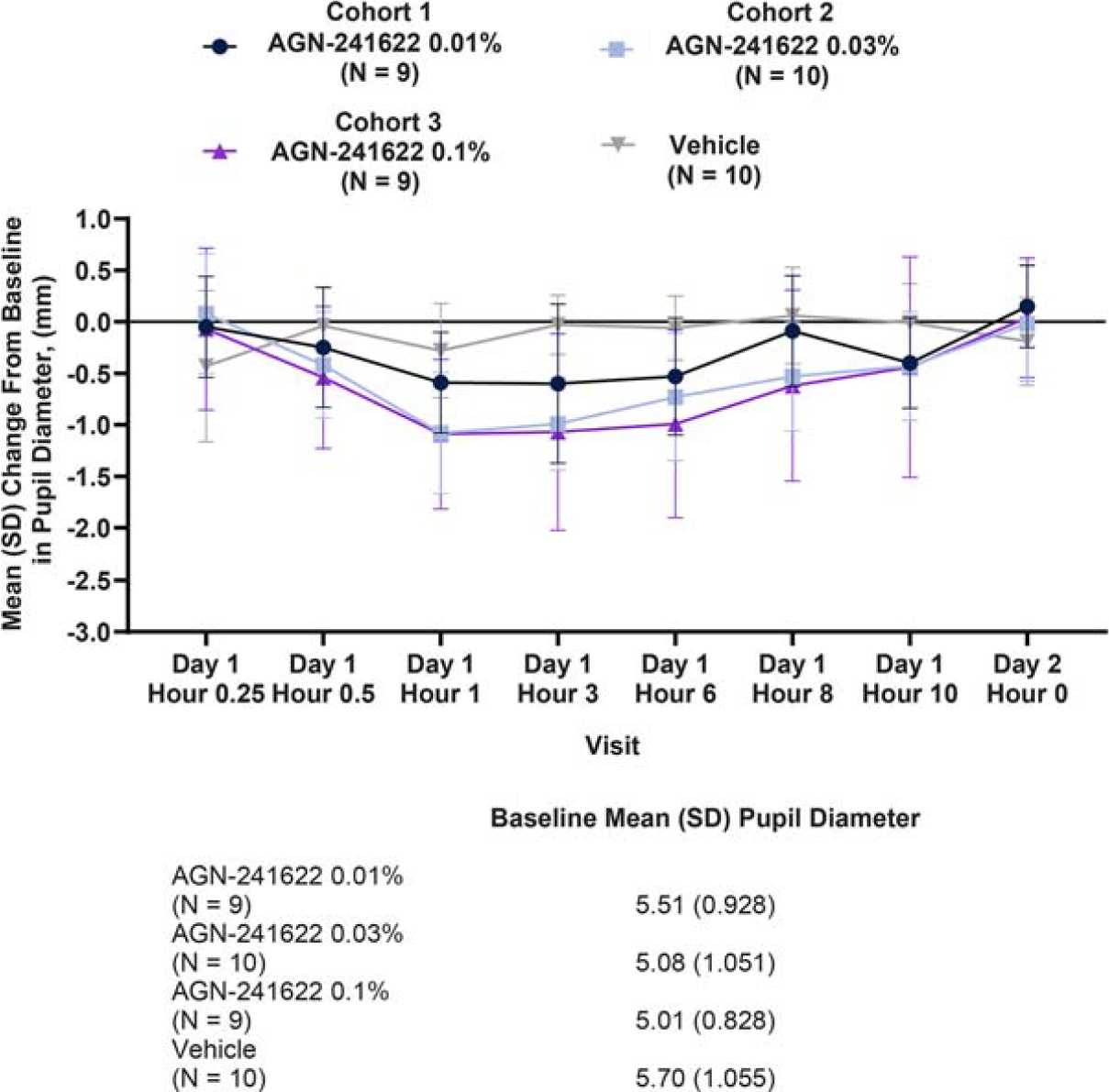
Stage 1 Mean (SD) Change From Baseline in Pupil Diameter (ITT Population). Change from baseline in pupil size assessed under mesopic conditions in the study eye. ITT, intent-to-treat; N, number of participants; SD, standard deviation.
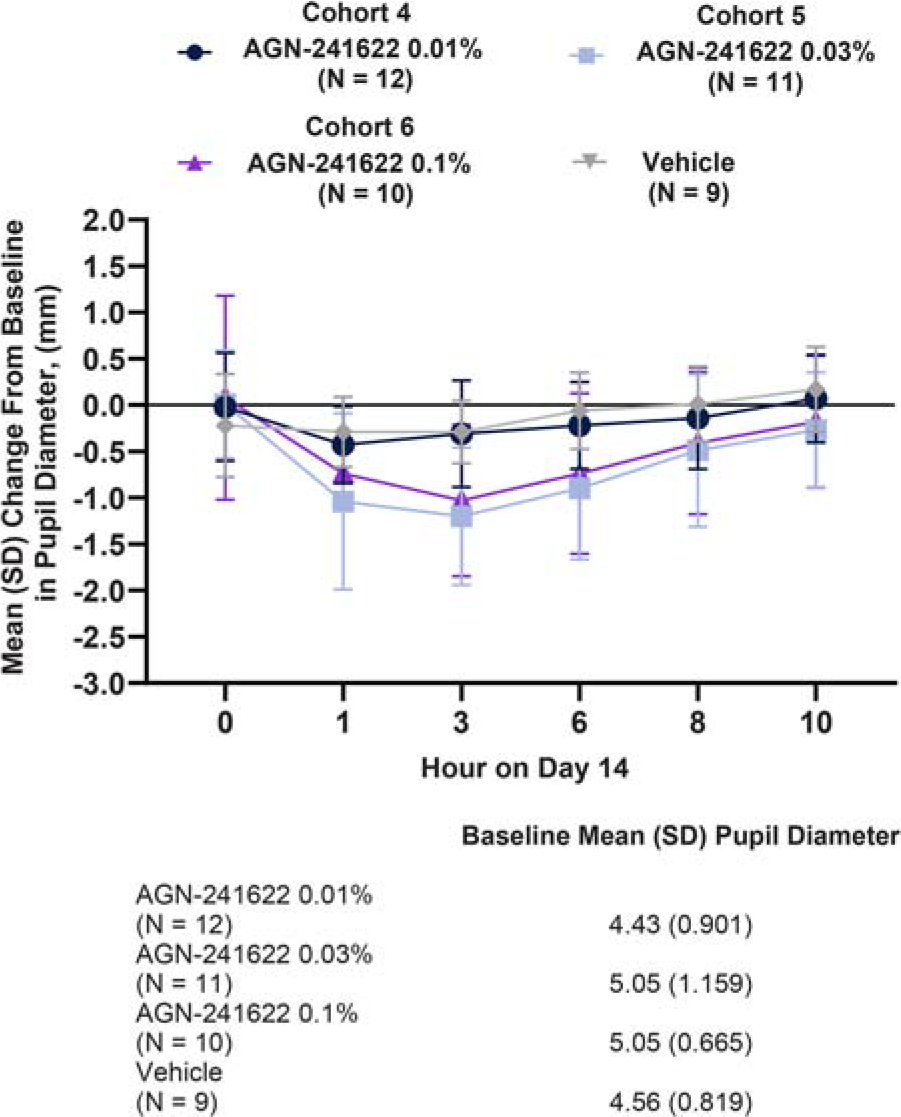
Stage 2a Mean (SD) Change From Baseline in Pupil Diameter (ITT Population). Change from baseline in pupil size assessed with distance correction under mesopic conditions in the dominant eye. ITT, intent-to-treat; N, number of participants; SD, standard deviation.
There was a higher proportion of responders with ≥3-line improvement in mesopic, binocular DCNVA in the AGN-241622 0.01% cohort compared with vehicle across most time points on day 14 (Table 3). Compared with vehicle, participants in the AGN-241622 0.01% cohort achieved the most gain in letters on day 14 with the peak occurring at hour 0.5 (mean [SD] change from baseline of 7.1 [4.6] letters) and continued at hour 1 (mean [SD] change from baseline of 7.1 [3.7] letters) (Fig. 4).
Stage 2a Proportion of Participants with ≥3-Line Improvement Mesopic DCNVA at Day 14 (ITT Population)
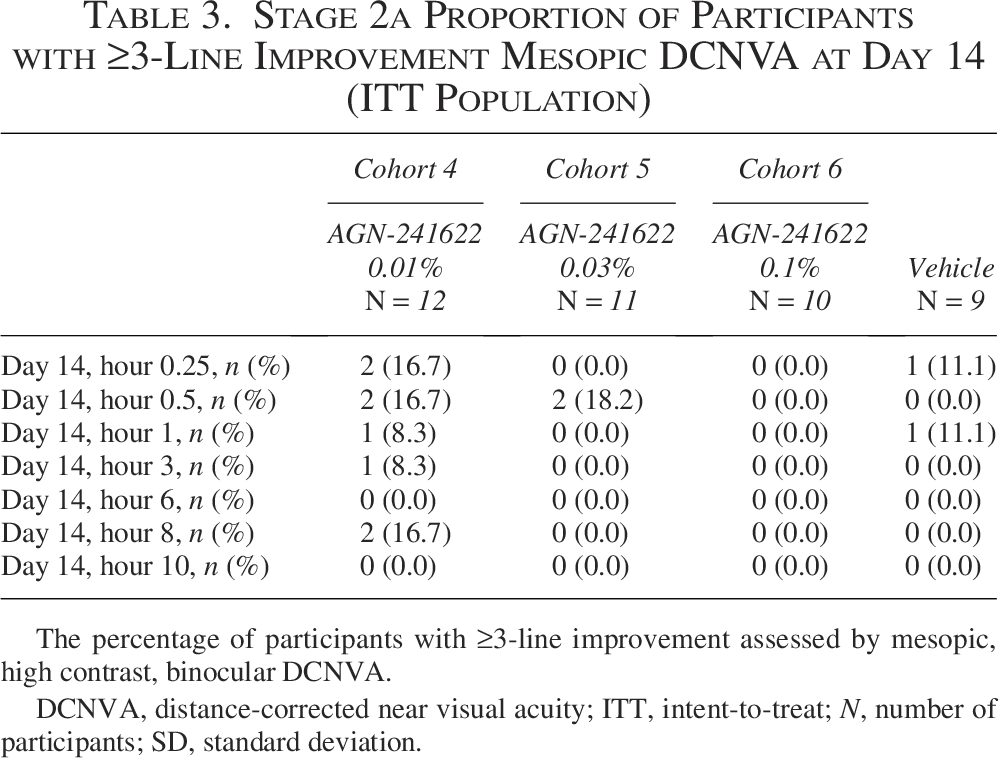
The percentage of participants with ≥3-line improvement assessed by mesopic, high contrast, binocular DCNVA.
DCNVA, distance-corrected near visual acuity; ITT, intent-to-treat; N, number of participants; SD, standard deviation.
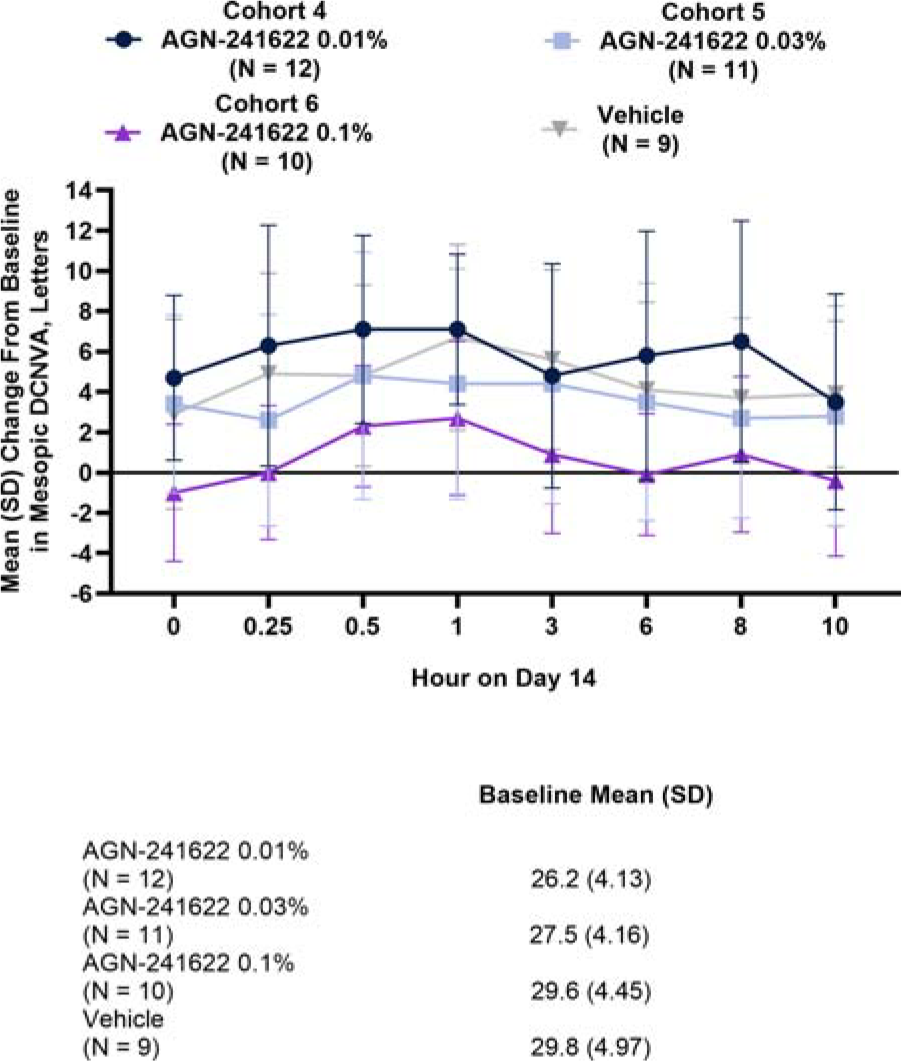
Stage 2a Mean (SD) Change From Baseline in Mesopic DCNVA at Day 14 (ITT Population). Change from baseline assessed binocularly under mesopic conditions. DCNVA, distance-corrected near visual acuity; ITT, intent-to-treat; N, number of participants; SD, standard deviation.
Safety
The safety population included 77 participants. In Stage 1, 40% (n/N = 14/35) of participants (AGN-241622 groups [n = 11] and vehicle [n = 3]) reported 1 or more TEAEs (Table 4). All reported TEAEs are listed in Supplementary Table S1. All TEAEs in Stage 1 were mild, and half of the events occurred in the AGN-241622 0.01% treatment group (n/N = 7/14). Of the ocular TEAEs in the AGN-2411622 0.01% group, the most frequently reported was increased lacrimation (50% [n/N = 4/8]). Eye irritation occurred in 25% of the participants in this group (n/N = 2/8]) and resolved without treatment. One participant in each of the AGN-241622 0.03% and vehicle groups also had increased lacrimation (Table 5). A low incidence of headache was observed across cohorts with 12.5% (n = 1), 11.1% (n = 1), and 22.2% (n = 2) of participants reporting mild headache in the AGN-241622 0.01%, AGN-241622 0.03%, and vehicle groups, respectively (Supplementary Table S1). No serious AEs or deaths occurred during the treatment period.
Overview of Adverse Events in Stage 1 and Stage 2a (Safety Population)
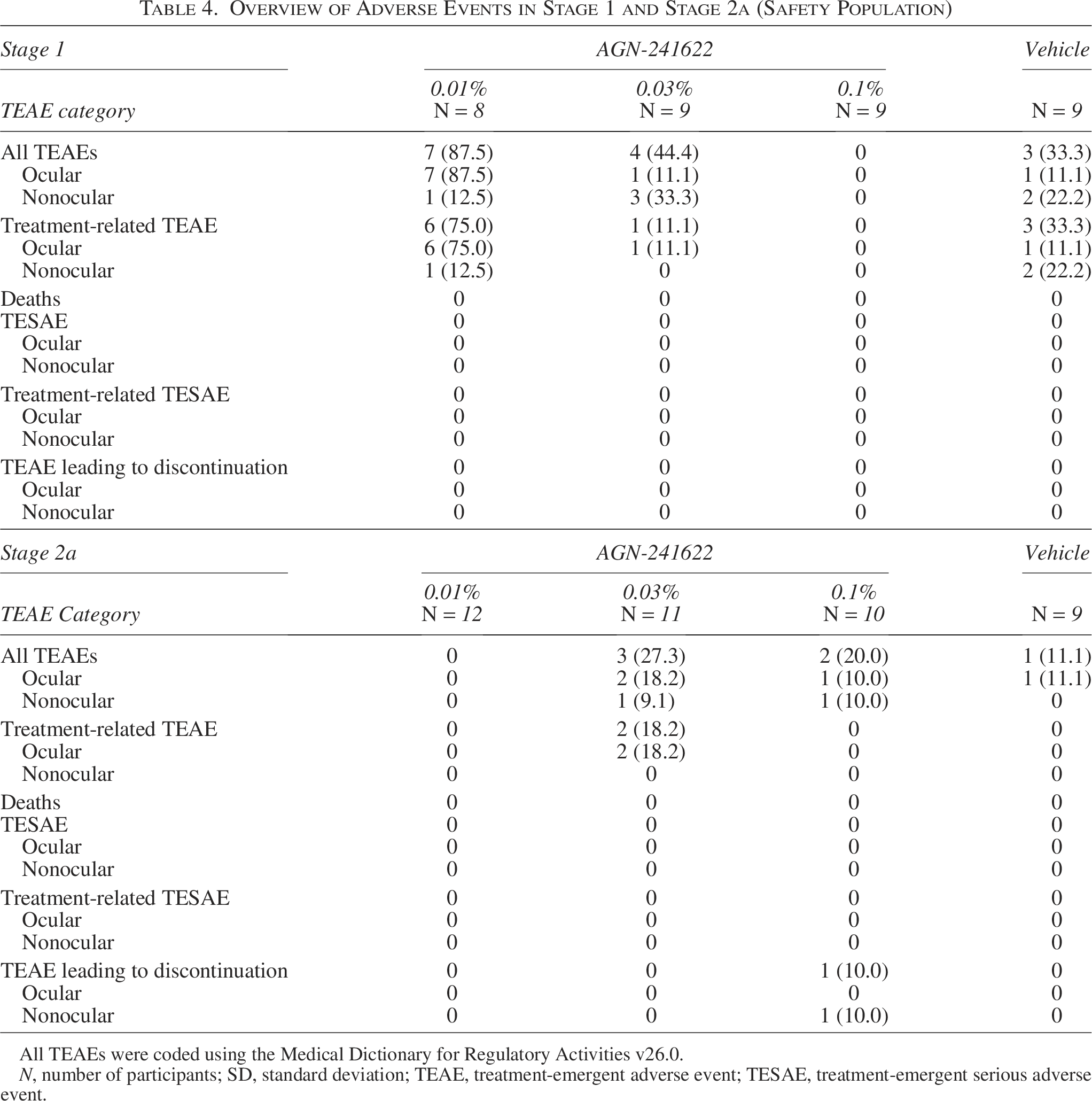
All TEAEs were coded using the Medical Dictionary for Regulatory Activities v26.0.
N, number of participants; SD, standard deviation; TEAE, treatment-emergent adverse event; TESAE, treatment-emergent serious adverse event.
Ocular Treatment—Emergent Adverse Events (Safety Population)
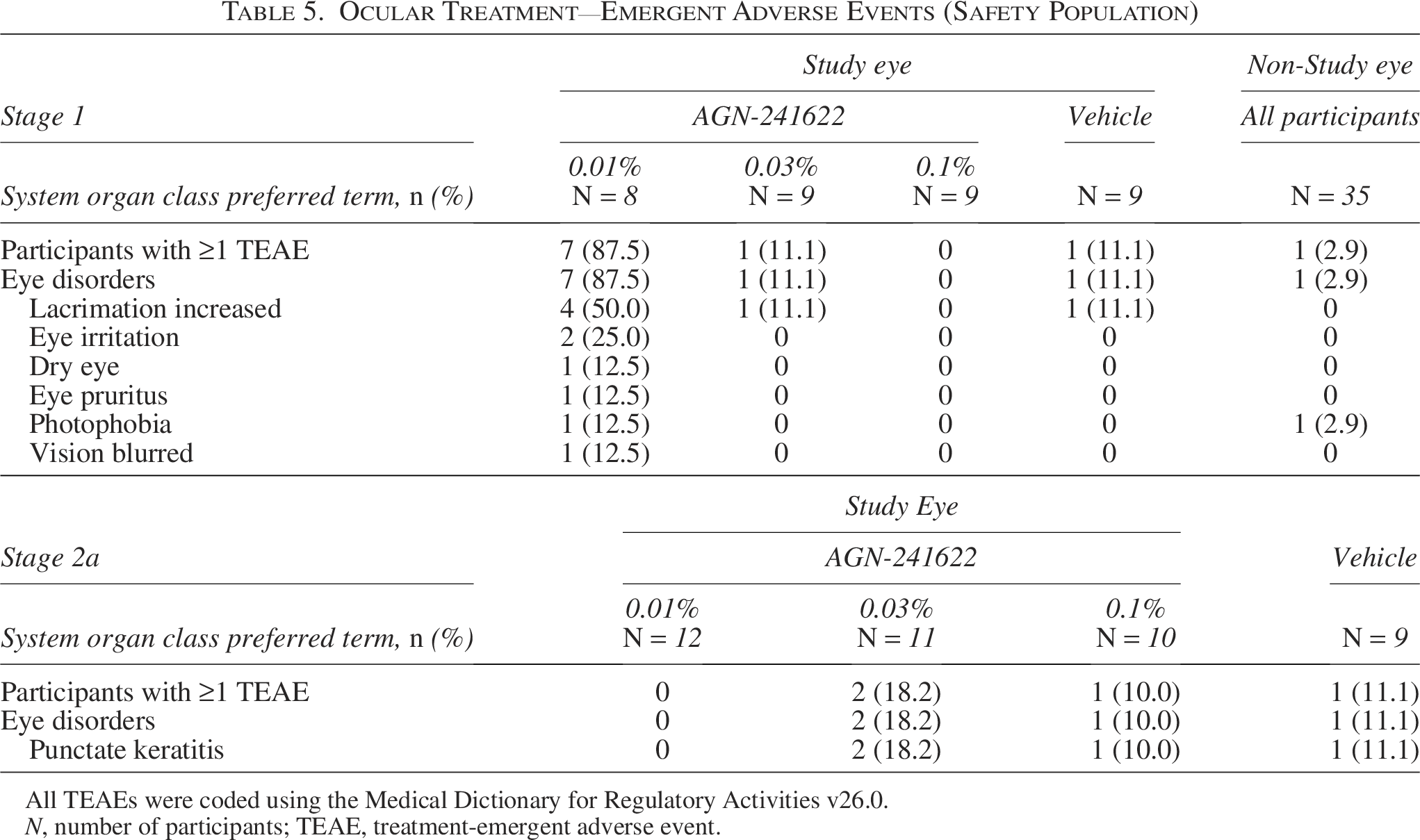
All TEAEs were coded using the Medical Dictionary for Regulatory Activities v26.0.
N, number of participants; TEAE, treatment-emergent adverse event.
In Stage 2a, 15.2% (n/N = 5/33) of participants in the AGN-241622 groups and 11.1% (n/N = 1/9) in the vehicle group reported 1 or more TEAEs (Table 4). No headaches were reported in Stage 2a; all TEAEs are reported in Supplementary Table S2. No deaths or serious AEs were reported during the treatment period. Punctate keratitis occurred in 0%,18.2%, 10%, and 11.1% of the AGN-241622 0.01%, AGN-241622 0.03%, AGN-241622 0.1%, and vehicle groups, respectively (Table 5). Two mild TEAEs of dermatitis in 1 participant from the AGN-241622 0.1% group led to study discontinuation, which resolved after treatment and was not related to AGN-241622. No clinically significant laboratory findings or physical findings related to AGN-241622 treatment were observed during the study. Overall, no safety concerns or trends were observed in either stage of the treatment period.
Discussion
In general, the results of AGN-241622 demonstrated a favorable safety profile and are comparable with other miotics approved or under investigation for presbyopia. 21 While headache is a common AE associated with miotic agents, participants reported fewer headaches with AGN-241622 compared with other treatments for presbyopia. 21 For example, Vuity (pilocarpine hydrochloride ophthalmic solution 1.25%, Allergan, an AbbVie company), the first eye drop approved for presbyopia in the United States, significantly improves near visual acuity without compromising distance vision with mild AEs, and 14.1% of participants in the Gemini 1 phase 3 trial reported headache.17,18 Results from the recently published CSF-1 pooled NEAR phase 3 studies of 0.4% pilocarpine hydrochloride reported headache in 6.8% of participants. 22 Moreover, the safety and efficacy of carbachol, and in combination with brimonidine, have been studied for the treatment of presbyopia and also reported mild to moderate AEs including headache and eye irritation. 23
In addition, modest efficacy was observed with AGN-241622 compared to studies of pilocarpine hydrochloride ophthalmic solution 1.25% and 0.4% pilocarpine hydrochloride.17,22 Results from the pilocarpine hydrochloride ophthalmic solution 1.25% Gemini 1 phase 3 trial demonstrated a 52.1% reduction at peak efficacy (−2.4 mm from 4.6 mm at baseline under mesopic conditions). 17 The maximum change from baseline in the pooled phase 3 studies of 0.4% pilocarpine hydrochloride occurred 1-h post-dose on day 8 (−1.23 mm compared with −0.06 mm in the vehicle group). 22 However, a peak reduction of 23.8% (−1.2 mm from 5.05 mm) was observed in the AGN-241622 0.3% and 20.4% (−1.03 mm from 5.05 mm) in the AGN-241633 0.1% cohorts on day 14 h 3. Similar to pupil response results, pilocarpine hydrochloride ophthalmic solution 1.25% showed a greater proportion of participants than AGN-241622 with DCNVA improvement of 3 lines or more. In the Gemini study, 41.6% of participants treated with pilocarpine hydrochloride ophthalmic solution 1.25% had DCNVA improvement of ≥3 lines. 17 In this study, peak efficacy occurred at hour 0.5 on day 14 with 16.7% and 18.2% of participants in the AGN-241622 0.01% and 0.03% cohorts demonstrating improvement of ≥3 lines. Although a pupil diameter reduction increased with increasing dose strengths of AGN-241622, a dose response was not observed in the improvement of DNCVA with increasing dose strengths.
Overall, presbyopia is associated with a decreased quality of life, and although several treatment options are available, they are not effective for all patients and have limitations.24,25 Eyeglasses and progressive lenses produce optical aberrations and can increase the risk of falls, especially in older patient populations.13,26 Treatments requiring surgical technologies may have surgical risks, the need for repositioning, or regression of effect, which have limited their widespread adoption.27,28 Miotics are a promising class of pharmacological treatments for presbyopia due to their ability to induce pupil constriction and increase the depth of focus and sharpness of near vision, which addresses the primary symptom of presbyopia.15,29,30 The investigation of miotics extends beyond pilocarpine and carbachol to include other cholinergic agents such as aceclidine, although these are still in preliminary stages of research. 15 The safety and efficacy profiles of miotics are critical areas of focus, as new formulations and delivery methods aim to minimize AEs while improving therapeutic benefits. Understanding individual variability in response to these treatments is important, as patient selection plays a substantial role in outcomes. 29 As research progresses, the long-term impact of miotic treatments in real-world settings will become clearer, potentially transforming the management of presbyopia and offering noninvasive, effective solutions for millions of affected individuals worldwide.
Limitations
While AGN-241622 was generally safe in healthy participants and participants with presbyopia, this was a first-in-human study, and the sample size was empirically determined to assess safety and efficacy.
Conclusions
In summary, results from the first standalone alpha agonist evaluated for the treatment of presbyopia demonstrated a favorable safety profile. Of 80 randomized participants, no serious AEs were reported with AGN-241622 treatment. All TEAEs were of mild severity, and there were no clinically significant laboratory or physical findings that were determined to be related to AGN-241622. In addition, AGN-241622 resulted in modest efficacy including pupillary reduction. A modest dose response in reduction of pupil diameter size was observed. However, a dose response was not observed in improvement in vision with increasing dose strengths.
Authors’ Contributions
Conception/design: S.L., A.N., M.R.R., and D.W. Data acquisition: S.M.E.-H., J.H.P., and D.W. Statistical analysis: J.Y. Data interpretation: S.L., A.N., J.Y., and M.R.R. Article review and approval: All authors.
Footnotes
Acknowledgment
Allergan, an AbbVie company, and the authors thank all study investigators for their contributions and the patients who participated in this study. Medical writing support was provided by Catherine Barone, PhD, of AbbVie. Editorial support was provided by Angela T. Hadsell, BA, of AbbVie.
Author Disclosure Statement
S.M.E.-H. has received honoraria and/or travel reimbursement from Aerie Pharmaceuticals, Allergan, an AbbVie company, Dompe, Novartis, and Sun Pharmaceutical Industries. J.H.P. received fees from Allergan, an AbbVie company, as a researcher on the study. D.W. consulted for Allergan, an AbbVie company, and Eyenovia and has received grant support from Aerpio, Allergan, an AbbVie company, Annexon, Dompe, Eyenovia, Mallinckrodt, Nicox, Novaliq, Novartis, SilkTech, and Santen. S.L., A.N., M.R.R., and J.Y. are employees of AbbVie and may hold stock and/or stock options.
Funding Information
Allergan, an AbbVie company, funded this study and participated in the study design, research, analysis, data collection, interpretation of data, reviewing, and approval of the publication. All authors had access to relevant data and participated in the drafting, review, approval, and decision to submit this article for publication. No honoraria or payments were made for authorship.
Data Sharing Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the United States and Europe and after acceptance of this article for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: ![]() then select “Home.”
then select “Home.”
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.