
Abstract
This article focuses on cutaneous hematopoietic neoplasms that are more likely to be encountered in the pediatric age-group and includes both lymphoproliferative and histiocytic disorders. The cutaneous hematologic disorders in children have a different epidemiologic profile to what is seen during adulthood. Although mycosis fungoides is the most frequent form of cutaneous lymphoma in adults, it is very rare in children. Because lymphoblastic leukemias and lymphomas are more frequent in the pediatric setting, cutaneous leukemic infiltrates are relatively common in this age-group. Similarly, histiocytic disorders are more common in children, particularly Langerhans cell histiocytosis and juvenile xanthogranuloma. Notably, the histiocytic disorders have undergone significant modifications on their nomenclature in the basis of the molecular characteristics that are present in them. A summary of the most frequent cutaneous hematopoietic disorders in children will be discussed further in this review.
Introduction
Like adults, the skin may be the first site of presentation for a hematopoietic neoplasm in a child or a site for involvement by advanced stage disease. A careful team-based approach that involves hematologists, dermatologists, and other specialists allows for an adequate and careful approach to the diagnosis of these neoplasms. A rapid morphologic interpretation of some of these tumors involves the use of cytologic touch-preparations, flow cytometry, fluorescence in situ hybridization (FISH), and conventional cytogenetics. It is capital to make a careful morphologic decision that can help decide what panel of immunohistochemistry (IHC) or molecular tools to use.
Four major histologic subtypes account for most non-Hodgkin lymphomas (NHLs) in the pediatric setting: Burkitt lymphoma (39%), diffuse large B-cell lymphoma (16%), lymphoblastic lymphoma (28%), and anaplastic large-cell lymphoma (10%). 1 Approximately 7% of pediatric NHL includes more unusual histologic subtypes, including cutaneous lymphomas. The epidemiologic aspects of cutaneous hematopoietic tumors in children are poorly studied, as most of them are derived from small case series that are highly biased by private consultation material from single institutions. 2 In the series by Fink-Puches et al. 3 (n = 69), approximately 35% of pediatric cutaneous lymphoma were accounted by mycosis fungoides (MF), another approximately 35% by CD30+ lymphoproliferative disorders (LPD; anaplastic large-cell lymphoma and lymphomatoid papulosis (LyP)), and 20% included marginal zone lymphomas (MZLs) and B-lymphoblastic lymphoma. The pediatric series by Boccara et al. 4 in France (n = 51) showed that nearly half of these tumors (47%) were composed by LyP, 27% by lymphoblastic lymphomas/leukemias and leukemic infiltrates by acute myeloid leukemia (AML), and approximately 10% included cases of EBV+ lymphoproliferative neoplasms and MF.
Some differences between adults and children are also present in the diagnosis of these diseases: while MF is the most common skin lymphoma in adults and children, a disproportionate number of the lesions in children have a particular clinical and immunophenotypic profile: clinical hypopigmented lesions and folliculotropic forms are much more prevalent in the pediatric setting. Most of the hypopigmented forms have a CD8+ phenotype. Invariably, all cases in children have a very indolent behavior. Similarly, leukemic infiltrates by lymphoblastic and myeloid leukemias are overrepresented in this age-group, as the diseases occur with a relative greater frequency in the pediatric setting. Histiocytoses with cutaneous presentation are more frequent in children as compared to adults.
This article focuses on cutaneous hematopoietic neoplasms that are more likely to be encountered in the pediatric age-group and includes both lymphoproliferative and histiocytic disorders.
Lymphoproliferative Neoplasms
Mature T-cell and NK-cell Lymphomas
Primary cutaneous ALCL, ALK−
Cutaneous anaplastic large-cell lymphoma (C-ALCL) is a primary cutaneous lymphoma composed of large and pleomorphic cells (indistinguishable from ALK+ ALCL) with diffuse expression of CD30 in the tumor cells (greater than 75%).5,6 The diagnosis is limited to those cases without a history of LyP or MF.7,8 Unlike ALK+ ALCL, C-ALCL is very rare in children compared to adults.9–14 In some cases of C-ALCL in childhood, an association with HIV infection has been documented. 9
Rapidly growing, asymptomatic, solitary, or multiple skin nodules/tumors with a tendency to ulcerate are the characteristic clinical presentation. 12 A neutrophilic variant (pyogenic) of C-ALCL contains a neutrophilic-rich infiltrate. Although the conventional variant does not have a predilection for immunocompromised individuals, the pyogenic variant is seen in those with HIV, 15 transplant recipients, 16 and hematologic malignancies including young individuals.10,17
Microscopically, ALCL has diffuse, cohesive sheets of large pleomorphic tumor cells, strongly CD30+, similar to ALK+ ALCL (Figure 1). Even among the conventional variants, a rich inflammatory infiltrate is present, which can lead to a misdiagnosis of an inflammatory process in the skin.
18
The tumors are frequently CD4+ and show expression of cytotoxic markers (TIA-1, perforin, and granzyme B). There is variable loss of T-cell antigens and, as opposed to systemic ALK+ ALCL, C-ALCL is usually negative for epithelial membrane antigen (EMA).
19
CD99 can also be positive.
9
The pyogenic variant can have a higher rate of CD8 and EMA (57%) expression, compared to the conventional form.10,20 Clonality is proven in the vast majority of cases by conventional T-cell receptor (TCR) studies. ALK− ALCL has shown convergent mutations and kinase fusions that lead to constitutive activation of the JAK/STAT3 pathway, and a subset with rearrangements at the locus containing DUSP22 and IRF4 in chromosome 6p25 tends to be relatively monomorphic, usually lack cytotoxic granules, and have been reported to have a superior prognosis, whereas a small subset with TP63 rearrangements is very aggressive.21–23
Primary cutaneous anaplastic large-cell lymphoma, ALK−. Psoriasiform changes of the surface epidermis are noted. There is a dense malignant infiltrate in the dermis with sparing of the epidermis. The tumor cells are very pleomorphic and large and have a background of acute inflammatory cells (neutrophils and eosinophils).
The clinical differential diagnosis for the pyogenic variant of C-ALCL includes pyoderma gangrenosum, pyoderma faciale (rosacea fulminans), Sweet syndrome, leishmaniasis, deep fungal infection, or pyogenic granuloma. Bacterial cellulitis has also been described as another differential diagnostic consideration. Among the neoplastic conditions, the main considerations include LyP (see discussion below) and tumor stage MF; the latter 2 disorders are best distinguished on a clinical background. Those with transformed MF have had a long-standing history of the disease and present with multiple tumors, whereas LyP has a history of self-remitting papules. Anaplastic lymphoma kinase (ALK) staining and CD30 are useful to rule out the possibility of cutaneous involvement by systemic ALK+ ALCL. 10
Mycosis Fungoides
MF is the most common form of cutaneous T-cell lymphoma in both adults24,25 and children.4,26–29 MF in children has a characteristic clinical appearance of hypopigmented patches and plaques that generally do not progress to the tumoral phase and has a CD8+ phenotype. 30 It has been reported in children as young as 3 years of age.3,27,31–34 The male to female ratio is nearly equal in the pediatric setting.27,31,34,35 Fink-Puches et al. 3 showed that MF represented almost 35% of all cutaneous lymphomas in individuals younger than 20 years of age.
Clinically, pediatric and adult MF is a protean disease that can potentially mimic numerous benign inflammatory dermatoses. In fact, a delay in the diagnosis can take several years, and numerous biopsies are often required to eventually establish or consider the diagnosis before clonality studies are performed. Ackerman raised concerns about the diagnosis of MF in the pediatric setting in the past: in their review of 106 cases, it was claimed that only 23 cases had information “sufficient” for a diagnosis of hypopigmented MF. 36 Indeed in 83 of those cases, there was no significant clinicopathologic correlation to confidently establish the diagnosis. MF also enters into the differential diagnosis of psoriasis, tinea corporis, pityriasis lichenoides, lichen aureus, atopic dermatitis, and the various hypopigmented dermatoses. Vitiligo and pityriasis alba are the 2 most frequent misdiagnoses in children.37,38 A recent case series from Castano et al. 26 reported that all cases (100%, n = 69) in children presented at the patch stage; almost all cases had hypopigmented lesions and most of these occurred in African-American children. The more frequent lesions in adults that consist of sharply demarcated patches with atrophy, and a cigarette paper appearance, are less common in children. An earlier study by Crowley et al. 27 of 58 patients with MF (younger than 35 years) showed that 17% presented with the tumor stage, and approximately 4% had generalized erythroderma. In another series of 46 cases, Heng et al. 39 reported that over 90% had hypopigmented lesion. The most common locations of presentation included the buttocks, trunk, and extremities (sun-protected areas). About 6% of patients have a solitary lesions. Rare cases of MF can present following organ transplantation. 40 Folliculotropic MF (FMF) presents with follicular papules occasionally with an erythematous base and follicular plugging and/or alopecia; this is the second most common clinical variant and seemingly occurs more frequently in individuals less than 40 years of age (8% of all variants).41–45 In FMF, the lesions are usually located in the head and neck region, with the presentation of plaques and/or tumors and intense pruritus. FMF has been associated with a worse outcome in adults and children unlike the more indolent and common patch/plaque lesions without FMF. 41 Less common clinical variants in the pediatric population include “granulomatous slack skin” or granulomatous MF,46–50 characterized by the development of areas of pendulous, lax skin in the major skin folds (especially axillae and groins). 51 Localized pagetoid reticulosis (Woringer–Kolopp disease) presents as a solitary, slowly growing erythematous and verrucous plaque on the extremities; rare cases have been reported in children.52–54 Sézary syndrome (SS) is a distinctive erythrodermic cutaneous T-cell lymphoma, characterized by pruritic erythroderma, generalized lymphadenopathy, and circulating malignant cells with cerebriform nuclei. It is a disease of adults, with only rare cases in the pediatric population.55–57 A rare clinical presentation of palmo-plantar keratoderma has also been described in children. 58
Morphologically, pediatric MF is histologically indistinguishable from that seen in adults (Figure 2). According to the series from Castano et al., the most frequent histologic findings include: lymphocytes in clusters, patchy lichenoid infiltrates, perivascular/periadnexal infiltrates, psoriasiform hyperplasia, papillary dermal fibroplasia, dermal melanophages, and Pautrier microabscesses. The infiltrate shows epidermotropism with tagging of cells along the dermal–epidermal junction.
59
The atypical lymphocytes show nuclear hyperchromasia and irregular and sometimes cerebriform nuclei. Distinctive perinuclear halos are seen surrounding the intraepidermal lesional cells. Admixed histiocytes, plasma cells, and eosinophils can be present. As the lesions progress clinically, so does the extent of the infiltrate. Large-cell transformation is defined as the presence of large cells (at least 4 times the size of a small lymphocyte) comprising more than 25% of the infiltrate or in nodular aggregates. Only rarely has such a phenomenon been described in lesions in children.60,61 In FMF, there is infiltration of the hair follicle epithelium with or without epidermotropism (epidermotropism is more frequently seen). The infiltrate involves the infundibulum of the hair follicle and at times deeper portions of the follicle. Follicular mucinosis may be an accompanying feature that can be better demonstrated by colloidal iron or Alcian blue stains. In addition, FMF is often accompanied by a syringotropic infiltrate.
41
However, a Dutch study revealed that interfollicular epidermotropism is actually rare.
45
FMF is not usually accompanied by intraepidermal Pautrier microabscesses.
62
Dermal eosinophilia can be prominent, particularly during the progression of the disease, and might be a manifestation of an autoimmune response to the keratin of the hair shafts in the dermis. In granulomatous MF, dense nodular and diffuse granulomas are present in the dermis, with or without epidermotropism, and with destruction of elastic fibers and elastophagocytosis. In the pagetoid reticulosis variant of MF, prominent pagetoid epidermotropism is noted. Such cases show a very impressive clinical response to radiation treatment (Figure 3).
Pediatric mycosis fungoides, hypopigmented variant. Ill-defined hypopigmented patches are present in the leg. Histopathologically, there is an atypical infiltrate with epidermotropism. Tagging of lymphocytes along the dermal–epidermal junction are seen. The lymphocytes are small, with hyperchromasia, irregular nuclear borders and perinuclear halos. Pediatric mycosis fungoides, pagetoid reticulosis variant. In this case, the infiltrate shows very extensive pagetoid epidermotropism. Lymphoma cells are CD4+ and CD8− and have loss of CD7.

The immunophenotype of pediatric MF is different from most of the adult forms (Figure 4): hypopigmented MF is a CD8+ cytotoxic T-cell lymphoma (whereas most cases in adults are CD4+).3,26–29,31,32,36,37,39,41,63–70 Also, CD30 expression is usually negative or reacts with only a few scattered cells (80% of cases are entirely nonreactive). Decreased expression of both CD4 and CD8 can also be seen in some cases of hypopigmented MF.71,72 Similarly, to adult MF, there is usually preserved expression of CD2 and CD5 with loss of CD7. A single rare case with coexpression of CD56 has been reported with an associated indolent clinical course.
73
In FMF, the infiltrate usually shows a predominance of CD4+ cells like the adult cases.
41
Granulomatous MF is also a disease of CD4+ T-cells. The genetics of MF and SS is less well understood in the context of the pediatric cases when compared to adult MF. When T-cell gene rearrangement studies have been performed in pediatric MF, TCR clonality was proven in approximately 64% of cases, and a polyclonal result was obtained in 16% of cases. These numbers appear to be lower when compared to the adult experience of 80% clonality which may reflect a relatively lower number of neoplastic cells in skin biopsies from children. However, sensitivity for TCR is lower in early lesions of MF (up to 60%).
74
Hodak et al.
41
revealed that monoclonality was demonstrated in only 43% of cases.
Pediatric mycosis fungoides, hypopigmented variant—IHC. The neoplastic cells are positive for CD3 and CD8. The CD4:CD8 ratio is inverted. There is aberrant loss of CD7.
The differential diagnosis of MF includes a number of inflammatory dermatoses: Pityriasis alba usually presents with hypopigmented macules and patches, situated in the face, but also on the trunk and arms. Histologically, there are very sparse lymphoid infiltrates, with very slight spongiosis, and without the dermal fibrosis seen in MF; vitiligo consists of depigmented macules and patches in localized, segmental, or widespread distribution. The lesions are usually on the face and distal extremities. Histologically, the findings are minimal, and if present, there is a subtle perivascular infiltrate of lymphocytes and few of them in the epidermis. As lesions evolve, there is a decrease in the numbers of melanocytes at the dermal–epidermal junction, which can be proven with the use of a MART-1 immunostain. Acral pseudolymphomatous angiokeratoma of children can also mimic pagetoid reticulosis. A dense lymphoid infiltrate beneath the epidermis, and thick wall vessels are seen. 75
Lymphomatoid Papulosis
LyP is a CD30+ T-cell LPD characterized by recurrent crops of papular lesions that usually prevail for 3 to 8 weeks and then resolve spontaneously. 76 The lesions are clinically “benign” appearing, but the histologic features suggest otherwise. LyP is the third most common type of cutaneous LPD in children.3,77 Boys are reported to have an earlier onset of LyP compared to girls. It is estimated that between 5% and 20% of those with LyP can develop a subsequent lymphoma, usually MF, Hodgkin lymphoma, or ALCL; this proportion appears to be smaller in the pediatric setting (approximately 9%).77–82
Clinically, LyP presents with erythematous papules and nodules that become hemorrhagic and necrotic after a few days with resolution as a depigmented scar.4,7,66,76–81,83–86 Lesions at different stages of evolution are characteristically present. In children, there is a clinical resemblance to pityriasis lichenoides, the most important disease in the differential diagnosis. The sites of predilection are the trunk and extremities, but other locations have been reported. 86 Regional LyP with crops of lesions located in a single anatomic region is seemingly more common in children.85,86 Nearly 50% of cases in children have more than 50 lesions at one point during their clinical course.
Several histologic variants have now been described in LyP: types A, B, C, D, E, and F. None of the histologic subtypes have any prognostic significance. The importance in differentiating the various subtypes is related to the differential diagnosis that each subtype entails. Many of these lesions have substantial histopathologic overlap: Type A LyP has a wedge-shaped dermal infiltrate composed of medium to large pleomorphic cells that are scattered throughout the infiltrate or arranged in clusters (Figure 5). The malignant cells can show features of Hodgkin-like cells, and there is a rich background of inflammatory cells. Ulceration, edema of the dermis, and focal vasculitic changes may be present; Type B LyP is characterized by an epidermotropic infiltrate reminiscent of MF; and Type C LyP has features identical histologically to C-ALCL, and clearly the clinical distinction is more important in a sense than the histologic findings. Cerroni et al. showed that most cases in children represented types A and C, with no examples of type B. In another pediatric series of 250 LyP cases in children, most cases were type A.
82
Type D LyP mimics aggressive epidermotropic CD8+ T-cell lymphoma histologically;
87
this variant expresses other cytotoxic markers such as granzyme and TIA-1. In the latter study, 4 of the 9 original patients were less than 25 years of age. Type E LyP is characterized by an angioinvasive and angiodestructive CD8+ cytotoxic T-cell infiltrate with CD30 coexpression; 2 of the 16 original patients were children.
88
Another study of 14 children with LyP revealed that 10 cases had a CD8+ cytotoxic phenotype; most cases were considered either type A or type C. Pierard et al.89,90 introduced the term follicular LyP (type F) to describe a subtype with a predominant perifollicular pattern of infiltration. A few cases of LyP (type F) have been reported in children.91,92
Lymphomatoid papulosis (LyP), type A. In LyP, there is a wedge-shaped infiltrate with only partial epidermal involvement. Malignant-appearing large and pleomorphic cells are present with a rich acute inflammatory background. The lesional cells are positive for CD30 and have significant loss of CD3.
The immunophenotype of types A and C is very similar with a uniform population of CD30+ cells, with frequent CD3 and CD4 coexpression. Typically, CD8 and CD56 are negative. CD15 is usually negative but cytotoxic molecules (TIA-1 and granzyme) are positive.93–95 Types D and E have a CD8+ phenotype. MUM1 is frequently expressed in all cases of LyP (but also in MF and C-ALCL). Some cases of LyP can also have a DUSP22 translocation. 96
Anaplastic Large-cell Lymphoma, ALK+
ALK+ ALCL represents approximately 10% to 30% of childhood lymphomas and is more frequent in the first 3 decades of life. There is a slight male predominance, and the majority of patients (70%) present with stage III or IV disease and B-symptoms. The skin is the most frequent extranodal site of involvement (present in 26% of cases). Other extranodal sites include bone, soft tissues, lung, and liver.97–100
ALK+ ALCL has a variety of morphologic presentations. All cases show a malignant population of large cells with eccentric horseshoe or kidney-shaped nuclei with an eosinophilic region near the nucleus, referred to as “hallmark” cells. In some variants, the hallmark cells can be small. Occasionally, the malignant cells can mimic Reed–Stenberg cells or their variants. There are 5 distinctive variants of ALK+ ALCL: common, small cell variant, Hodgkin-like pattern, lymphohistiocytic pattern, and combined forms.101–105 Any of these patterns may present in the skin (Figure 6). The immunophenotype of ALK+ ALCL includes CD30 and ALK expression; the pattern of CD30 expression is both Golgi and cytoplasmic. Most cases are positive for EMA, TIA-1, Granzyme B, and perforin, and most show loss of T-cell antigens such as CD3, CD5, and CD7. Some cases can be null for all T-cell markers. CD43, CD2, and CD4 are more frequently positive. Certain staining patterns of ALK expression correlate with specific rearrangements of the ALK gene. However, the most common cytogenetic rearrangement represents the t(2;5) translocation creating a fusion transcript involving the ALK and NPM genes (nucleophosmin). Such translocation is associated with the classic cytoplasmic and nuclear staining pattern of ALK IHC.
Cutaneous anaplastic large-cell lymphoma ALK-positive. In this case, the malignancy manifested first on the skin. The clinical lesion had the appearance of a pyogenic granuloma. An ulcerated tumor is present. The malignant cells show features of “hallmark” cells. Many mitotic figures are present. The neoplastic cells are positive for CD30, ALK, EMA, and CD4.
Lamant et al. 106 suggested a possible relationship between insect bites and the development of ALK+ ALCL; this series of 5 patients had recent arthropod bites and developed nodal disease in the area of the skin lesions. Two of the skin biopsies revealed the presence of ALK+ cells. A complete remission was obtained after chemotherapy in all but one case who developed progressive disease and died. More recently Oschlies et al. 104 described a series of 6 children, within the context of 487 patients enrolled in the Anaplastic Large-cell Lymphoma-99 trial with disease limited to the skin; these patients had complete remissions on follow-up, and most were treated with surgical excision only. In all cases but one, the pattern of ALK staining was nuclear and cytoplasmic, and FISH was positive for the ALK-NPM translocation. Clinically, in 5 out of 6 cases, lesions were solitary (maculopapules or nodules) and 1 patient had multiple lesions. Previous isolated case reports of cutaneous presentations of ALK+ ALCL have been described.107–109 Despite such reports, most cases of ALK+ ALCL have associated systemic disease and require full staging.
Small- to Medium-sized CD4+ T-cell LPD
Small- to medium-sized CD4+ T-cell LPD is known to present in children, but in a minority of cases. 110 This putative disorder remains in a provisional category in the World Health Organization (WHO) classification. 111 The terminology in the revised classification has been modified from “lymphoma” to “LPD” to reflect this uncertain malignant potential, designating these cases as primary cutaneous CD4+ small/medium T-cell LPD. The clinical behavior is almost always indolent, with most patients presenting with localized disease. Systemic disease is rare, and conservative local management is sufficient in most patients. It has been suggested that this may represent a limited clonal response to an unknown stimulus, not fulfilling criteria for malignancy.23,112,113
All cases presenting to date in children have a solitary lesion, usually a papule, nodule, or plaque in the head and neck region. The infiltrate consists of a nodular, diffuse, and interstitial infiltrate of lymphoid cells with sparing of the epidermis. There is expression of CD4, but a rich background of B-cells is also encountered with the expression of germinal center markers (BCL-6, CD10, PD1, and ICOS).102,114,115 CD30 is usually negative.
Aggressive CD8+ Epidermotropic T-cell Lymphoma (Berti’s Lymphoma)
The original series of primary cutaneous epidermotropic CD8+ T-cell lymphoma had a single case in a child. 116 Kikuchi et al. 117 subsequently described a second case in a 6-year-old girl with disseminated lesions. A multicenter study of 30 cases included 2 cases in individuals younger than 25 years. The median survival is approximately 12 months. 118 Patients usually present with papulonecrotic lesions in the extremities. Morphologically, these lesions typically reveal an acanthotic epidermis with prominent epidermotropism and keratinocytic necrosis. The immunophenotype includes TCRβ, TIA-1, and CD8 expression.
Primary Cutaneous γδ T-cell Lymphoma
This extraordinary rare disorder has been reported in children, but it is basically an adult condition.4,119–124 Merrill et al. 125 reported the occurrence of primary cutaneous γδ T-cell lymphoma (CGDTCL) in 3 individuals before the age of 25 years, including a 1-year-old child. The lesions are usually found on the extremities and trunk. The malignant infiltrate is accompanied by epidermal necrosis with frequent involvement of the subcutis. Vasculitis and ulceration can be seen, and hemophagocytosis is also present. The most common immunophenotype includes CD3+, CD4−, CD8−, and CD56+/−. The identifying feature is the presence of TCRγ expression. An important differential diagnostic consideration is LyP since some cases in children can have TCRγ expression. The distinction between LyP and CGDTCL is an important one since the former is an indolent disease and the latter requires systemic chemotherapy and bone marrow (BM) transplant. The previously reported cases of subcutaneous panniculitis-like T-cell lymphoma (SPTCL) with a γδ phenotype have now been reclassified as CGDTCL. 126
Subcutaneous Panniculitis-like T-cell Lymphoma
SPTCL is more common in women and has a predilection for younger individuals with approximately 20% of cases in people younger than the age of 20 years. Numerous cases have now been reported in children.127–132 A subsequent study of cutaneous lymphomas in children noted that SPTCL was rare with only 3.4% of cases in this age-group.
66
The lesions are nodules/tumors or cutaneous plaques, which can vary in diameter from 1 to 20 cm or more. Areas of lipoatrophy are seen after resolution of the nodules. Hemophagocytosis is present in 17% of cases whose presence is associated with a poor outcome with death in more than 50% of cases; the poor prognosis is relatively common in T-cell lymphomas with a panniculitic presentation and the γδ phenotype or among cases in children (17% αβ vs 45% γδ).126,133 Gau et al.
134
reported SPTCL with hemophagocytosis occurring in a brother and sister, with an interval of 11 years between their diagnoses. A dense, nodular, interstitial, or combined lymphoid infiltrate showing adipotropism with a predilection for the subcutaneous lobule mimicking a lobular panniculitis is seen in virtually all cases (Figure 7).4,66,127,128,135–147 The immunophenotype reveals CD3+, BF1+, CD8+, and cytotoxic molecules, such as TIA-1, perforin, and granzyme B. Cases with γδ expression are now reclassified as CGDTCL. An important differential diagnosis of SPTCL is lupus panniculitis which can present in children.
148
In contrast to SPTCL, lupus panniculitis presents with subcutaneous fibrosis, frequent clusters of plasmacytoid dendritic cells (CD123+), reactive germinal centers, and absence of the phenotype of SPTCL.
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL). There is a diffuse infiltrate in the adipose tissue that simulates a lobular panniculitis. Adipocyte rimming of the neoplastic cells is noted. The infiltrate spares the epidermis and superficial portions of the reticular dermis. Courtesy of Path Presenter.
Systemic Chronic Active Epstein–Barr Virus Infection of T-cell or NK-cell Type
This rare systemic EBV+ polyclonal, oligoclonal, or monoclonal T-cell or NK-cell LPD manifests with variable clinical severity. It was originally defined as a severe illness with a duration of more than 6 months, after a primary EBV infection with continued high titers to EBV and evidence of major organ involvement (pneumonia, BM aplasia, uveitis, lymphadenitis, hepatitis, and splenomegaly). EBV by in situ hybridization (EBV-encoded RNA [EBER]) expression has been used to document EBV in the infected tissues. 149 The updated criteria also allow for the diagnosis in the presence of symptoms that last for longer than 3 months and have high levels of EBV viremia.23,150,151 Although the affected cells are typically T- or NK-cells, rare cases of B-cell derivation can occur. 152
The disease has a strong predilection for individuals of Asian or South American descent, and most notably occurs in children and adolescents.153–156 Less frequently, chronic active Epstein–Barr virus (CAEBV) can occur in middle age or older adults with prolonged fevers, hepatosplenomegaly, anemia, thrombocytopenia, lymphadenopathy, and cutaneous manifestations. The latter are typically in the form of mosquito bite allergy (33%), rash (26%), and hydroa vacciniforme (HV)-like manifestations. Most patients have high antibody titers (EBV VCA IgG, EBNA), as well as high EBV viremia.150,152,155–157
The cutaneous findings will be discussed with the HV-like and mosquito bite allergic disorders. In other sites, such as the lymph nodes, CAEBV is characterized by paracortical hyperplasia, a polymorphous infiltrate, and a rich background of inflammatory cells, that include plasma cells and granulomas at times. Some cases are accompanied by associated hemophagocytic lymphohistiocytosis (HLH), best demonstrated in BM, lymph node, and liver biopsies.155,158 Numerous EBV+ cells are seen. When monotonous sheets of EBV+ tumor cells are noted, such cases are best classified as systemic EBV+ T-cell lymphoma (ENKTL) of childhood or aggressive NK-cell leukemia (ANKL). 159
The prognosis of CAEBV is variable, with some patients experiencing a relatively indolent clinical course, while others succumb to the disease, especially in the presence of HLH. The median survival is approximately 70 to 78 months, and adverse markers of clinical outcome include late onset (>8 years of age), thrombocytopenia, and T-cell infection (5-year survival is approximately 60% for T-cell disease and 87% for NK-cell disease).150,152,158 T-cell clonality is sometimes associated with a more aggressive clinical course. 155
Severe Mosquito Bite Allergy
The cutaneous manifestations of CAEBV infection may be precipitated by mosquito bites or vaccination.160,161 Severe mosquito bite allergy (SMBA) is very uncommon, and most cases occur in individuals from Asia and Mexico.162–170 This disorder is not a hypersensitive reaction in the classic sense, but is rather a cutaneous manifestation of CAEBV of NK-cell lineage, with oligoclonal or monoclonal populations of NK-cells. 171
Most patients are less than 20 years of age, with a median age of 6.7 years. 171 Erythematous papules, macules, and plaques can subsequently evolve into bullae with ulceration and eventual scarring. Systemic symptoms are also common and include fever, lymphadenopathy, and liver dysfunction.172,173 Recovery occurs when the systemic symptoms resolve until the next mosquito bite. Vaccinations can also sometimes elicit a similar reaction. 174 High levels of IgE, high EBV viremia, and NK-cell lymphocytosis (80%) are present in SMBA. 160 The skin shows epidermal necrosis with ulceration. Marked dermal edema and infiltration by neutrophils and a dense lymphoid infiltrate are noted. Vasculitis is present in the center with karyorrhectic debris and extravasated red blood cells in the dermis. The lymphoid infiltrate is composed of a mixture of CD4+, CD8+ T-cells, and NK-cells. EBV+ cells represent a minority of the infiltrate (<10%).
The prognosis is variable: some patients have an indolent course, with chronic protracted cutaneous manifestations, while others may develop fulminant HLH or progress to lymphoma, such as aggressive NK-cell leukemia. 174
Hydroa Vacciniforme-like Lymphoproliferative Disease
This chronic EBV+ LPD of childhood has a risk of progression to a systemic lymphoma. 151 Classic HV is a rare, intermittent ultraviolet light-induced vesiculopapular and scarring eruption, that typically remits after adolescence.175–177 Systemic symptoms are not usually present in HV. The estimated prevalence of HV is approximately 0.34 cases/100,000. 175 In some cases, the lesions can occur in sun unexposed areas. Later studies have shown an association between HV and clonal proliferations of T-cells; the term HV-like lymphoma (HVLL) was introduced in the WHO classification in 2008. 178 However, because of the inability to predict which patients will have an indolent course, and which ones will develop overt lymphoma, the term HV-like lymphoproliferative disease (HV-LPD) has been proposed and will be introduced in the next edition of the WHO monograph. 23 Other terms that have applied to describe HVLL in the past included edematous scarring vasculitic panniculitis, angiocentric cutaneous T-cell lymphoma of childhood, hydroa-like cutaneous T-cell lymphoma, and severe HV. It has been proposed that certain populations (eg, Asian and South American) have a risk for development of lymphoma, while in others (North American and European), the disease has an indolent course.175,179 Recent clinical studies suggest that HV-LPD can be successfully treated with immunomodulators (thalidomide, 180 antivirals, 181 interferon, 182 etc) rather than systemic chemotherapy, as the latter has been associated with a higher mortality among these patients. 183
Most cases of HV-LPD are seen in children and adolescents from Asia, Native Americans from central and south America (Peru, Guatemala, and Bolivia), and Mexico and rarely occur in adults.176,179,183–190 In the Peruvian studies, approximately 50% of cases are found in the southern region of the country, correlating with a higher prevalence of early EBV infection in that area. The median age at diagnosis is 8 years. The male to female ratio is 2.3:1. Males with HV may have a later onset and longer duration than females. Although the disease presents in children, some patients can have persistent symptoms into adulthood.
Classical HV is characterized by a sporadic, itchy erythematous eruption in sun-exposed areas that occurs shortly (minutes to hours) after sun exposure. The eruption progresses through different stages: papules, vesicles, crusts, and eventually vacciniform (pox-like) scars after several weeks (Figure 8). Severe forms of the disease can have conjunctival and corneal ulceration and scarring. Marked facial edema is common in the more aggressive forms, associated with unilateral or bilateral eyelid compromise. Periorbital swelling can be the first manifestation of the disease.175,176,185 The lesions can become chronic (lasting months to years) and are prone to recurrence, often characterized by temporal heterogeneity, from papules with crusts to well-formed scars in the same area.
175
When extracutaneous involvement occurs (hepatosplenomegaly, lymphadenopathy, and BM infiltration), those cases are best classified as ENKTL of childhood. Fever, weight loss, and asthenia can be present. Elevations of lactate dehydrogenase and liver enzymes are noted in one-third of the patients.183,191,192
Hydroa vacciniforme lymphoproliferative disorder. A crusted popular rash is present in sun-exposed areas. A large area of epidermal necrosis is noted. There is a superficial and deep, angiotropic and perifollicular infiltrate. The infiltrate is composed of medium- to large-sized cells. Angionecrosis is proven by the presence of fibrinoid changes in the vessel walls.
In HV-LPD, the lymphoid infiltrate is composed of small- to medium-sized hyperchromatic cells centered in the dermis with a perivascular/periadnexal distribution, and with associated epidermal necrosis (Figure 8). Spongiosis and intraepidermal vesicles are also seen. In the more aggressive forms of the disease, the cells can show significant atypia, and somewhat similar morphology to ENKTL. Mitotic figures are infrequent. Epidermotropism without Pautrier microabscesses is a common feature. Fully developed lesions are deeply infiltrative into the adipose tissue, but without significant rimming of the adipocytes as seen in SPTCL. Angiotropism with or without angionecrosis and fibrinoid changes can be seen.179,183,186,188,189
The EBV-infected cells are cytotoxic T-cells (CD3+, CD8+, TIA-1+, granzyme B+, and perforin+) or NK-cells (CD56+) (Figure 9).179,183,186,189 A minority of cases can be CD4+ or CD4−/CD8−.193–195 CD30 is occasionally positive, but LMP-1 is negative. The number of EBV+ cells is variable. The Ki-67 index is variable and is very low in some cases or as high as 50% in others. Expression of CD5, CD7, CD43, and CD25 is variable. CD57 is negative. The T-cells can be positive for either TCRαβ or TCRγδ. Clonal rearrangements of the TCR can be demonstrated. Deletion of the long arm of chromosome 6 has been documented.
196
Hydroa vacciniforme lymphoproliferative disorder—IHC. The infiltrate is positive for CD3 but shows aberrant loss of CD5. CD7 appears to be retained. Many CD56+ cells are seen. HV-LPD shows a cytotoxic phenotype that is best proven by a granzyme B immunostain. EBER is positive in the lesional cells.
The differential diagnosis includes other NK/T-cell LPD, such as ANKL and ENKTL. In the latter entities diffuse sheets of atypical CD56+, EBER+ cells are noted. Some cases of ANKL can also show skin involvement. 197 SPTCL can have overlap in the immunophenotype, being composed of CD8+ cells. However, SPTCL is negative for CD56 and lacks epidermotropism and EBV expression. Primary cutaneous γδT-cell lymphoma (PCGDL) can also be CD56+ but is more typically negative for EBV, presents in older adults, and the location is more typically truncal and in the extremities.
Extranodal NK/T-cell Lymphoma
Extranodal NK/T-cell lymphoma, nasal type, is a mature NHL derived from NK-cells (more frequently) and sometimes T-cells with extranodal (frequently upper aerodigestive) infiltration by EBV-infected cytotoxic lymphocytes with angioinvasion and necrosis. 151 In the United States, ENKTL is very rare, accounting for fewer than 1% of NHL. The disease is more prevalent in areas of Mexico, South and Central America, and Asia where it represents nearly 6% of all NHLs, and in certain areas, up to 22% to 44% of all NHLs.198,199 Overall, approximately 150 cases of ENKTL have been reported in children.200,201,132 The median age of presentation is 13 years, with a male to female ratio of 3.25:1.
ENKTL shows a strong association with EBV infection, irrespective of the patient’s origin, which supports a direct pathogenic role of the virus in lymphomagenesis. The disease presents in extranodal sites, most often in the upper aerodigestive tract (70%), including the nasal cavity (by far the most frequent site), orbital soft tissue, paranasal sinuses, and palate (Figure 10). Some cases can present in extranasal cavity sites, but lymph node involvement is uncommon. Most often, there are signs of local tumor infiltration: ulceration, perforation of the nasal septum (in the past, many of these cases were called midline lethal granuloma
202
), and epistaxis are frequently seen.
151
B-symptoms such as fever, weight loss, malaise, and night sweats are also common (65% of pediatric cases) and are the second most common clinical presentation of ENKTL in children. ENKTL can also be associated with hemophagocytosis which worsens the prognosis.
203
Dissemination to the skin, gastrointestinal tract, testis, or cervical lymph nodes can also be present, while BM involvement is relatively uncommon. Skin involvement is present in 10% of cases of ENKTL and manifests as nodules that ulcerate and often have a necrotic center, erythematous maculopapules, cellulitis, and abscess-like swelling.
Extranodal NK/T cell lymphoma, nasal type (ENKTL). The infiltrate is dermal-based and shows a much more pleomorphic character compared to HV-LPD. Angiotropism with angionecrosis is noted. Many large and pleomorphic cells are seen. The malignant cells are positive for CD56, and all of them are EBER-positive.
The survival rate is 30% to 40%,204,205 but in recent years, it has improved with more intensive therapy including up-front radiotherapy and the steroid (dexamethasone), methotrexate, ifosfamide,
ENKTL has an angiocentric and angiodestructive growth pattern with coagulative necrosis, admixed apoptotic bodies, and fibrinoid change in blood vessels, even in the absence of angioinvasion (Figure 10).211,212 Pseudoepitheliomatous changes are present in some cases. The tumor cells can exhibit a marked range in cell size. The nuclei are round or folded and have coarse or vesicular chromatin and small inconspicuous nucleoli. Mitotic figures are frequently seen, in addition to apoptosis and karyorrhectic debris. Cytoplasmic granules are seen in some cases. Areas of geographic necrosis can also be present. Background inflammatory cells are variable and include plasma cells, histiocytes, and neutrophils. In the skin, focal epidermotropism is seen on occasion. When the adipose tissue is affected, necrosis and rimming of the adipocytes by the malignant cells is noted, which can mimic the findings present in CGDTCL and SPTCL. Permeation of the tumor cells into the skeletal muscle with associated myonecrosis can also be present.151,197
ENKTL originates from T or NK-cells with a cytotoxic phenotype and expression of cytotoxic granules (granzyme B, TIA-1, and perforin)213–216; however, most cases are derived from NK cells and are CD56+ and CD56− for surface CD3 expression yet positive for cytoplasmic CD3 subunits including CD3ɛ and CD3ζ. As such, ENKTL is positive for CD3 when polyclonal anti-CD3 antibodies are applied. Most cases are positive for CD2, CD43, CD45RO, HLA-DR, CD25, CD7, and FAS (CD95) and negative for CD4, CD5, CD8, TCRγ, βF1, CD16, and CD57.217,218 CD30 is positive in 20% to 40% of cases, particularly in cases with a rich large-cell component. 200 The Ki-67 proliferation index is typically very high (>50%), even in the presence of small cell-predominant tumors. In situ hybridization studies for EBER is positive in virtually all cases.
The TCR and immunoglobulin heavy chain (IGH) genes are in germline configuration in ENKTL derived from NK cells. Clonal rearrangements of the TCR genes are detected in 10% to 40% of cases, particularly in those of T-cell origin.219,220 Recent studies have demonstrated activation of the JAK-STAT signaling pathway in ENKTL. 151
The differential diagnosis of ENKTL in the skin includes other aggressive types of cutaneous lymphoma. PCGDL is often CD56+ but usually lacks expression of CD5, CD4, and CD8. As opposed to ENTKL, epidermotropism is more frequently found, and the lesions show strong expression of TCRγ. Rare cases are double positive for βF1, and some cases of PCGDL are TCR silent. EBER is only rarely expressed (<5% of cases). 120 MF can rarely be CD56+221,222 and, as opposed to ENTKL, shows more prominent epidermotropism and frequent formation of Pautrier microabscesses. Most cases of MF are positive for βF1 and more typically CD4+. Other entities included in the differential diagnosis are other EBV-associated T-cell or NK-cell LPDs such as EBV+ peripheral T cell lymphoma, not otherwise specified (PTCL, NOS), CAEBV infection, systemic EBV+ T-cell lymphoproliferative disease of childhood, and HV-like LPD, which also occurs predominantly in children. Aggressive NK-cell leukemia can also be included in the differential diagnosis and shares similar immunophenotypic features to ENKTL. However, skin involvement is relatively uncommon in the disease, allowing in the clinical distinction.
The distinction of PTCL, NOS from extranodal NK/T-cell lymphoma (ENKL) is typically based on the EBV status, following the recommendations of the WHO classification. ENKL is invariably EBV+, while PTCL, NOS in the setting of CD56 expression is EBV−. However, a recent study reported 7 cases of EBV− aggressive NK-cell leukemia, 3 of which had cutaneous involvement upon presentation. 223 The EBV− cases had an aggressive clinical course, similar to that of EBV+ aggressive NK-cell leukemia. Blastic plasmacytoid dendritic cell neoplasms (BPDCNs) also are CD56+ and also occur in children.224,225 Morphologically, BPDCNs are composed of medium-sized cells, with fine nuclear chromatin and blastic appearance. These lesions typically lack the angiotropism and angiodestruction present in ENKTL. Immunophenotypically, BPDCN is positive for CD123, BDCA2, TCL-1, and CD4 and show variable terminal deoxynucleotidyl transferase (TdT) expression, while negative for CD3 and EBV; these markers are negative in ENKTL.
Cutaneous B-cell Lymphomas/Leukemias
Marginal zone lymphoma (MZL)
is a low-grade B-cell NHL composed of small, monocytoid, and lymphoplasmacytoid cells and mature plasma cells.226,227 Many cases are associated with infectious etiologies, such as Helicobacter pylori (in gastric mucosa-associated lymphoid tissue [MALT]), Borrelia burgdorferi (in cutaneous cases only in Europe), and Chlamydia (ocular MALT) with site specificity. More recently, IgG4 expression has been shown in cutaneous cases with prominent plasmacytic differentiation. 228 Primary cutaneous MZL (PCMZL) is extraordinarily rare in children. A series from Kempf et al. 229 included a total of 3 cases, mostly in teenagers. In a study of cutaneous lymphomas in individuals younger than 20 years, Fink-Puches et al. showed a total of 7 cases of MZL. There is a slight predominance in girls compared to boys.230–234 The first case of the disease reported in the United States was found in association to chronic antihistaminic use. 232 Rare cases in association to medications for attentional deficit disorders have been seen. 229 Cases in relationship to medications use usually resolve upon cessation of the medication. More recently, Amitay-Laish et al. 235 reported a series of 11 cases of pediatric PCMZL; none of the patients had progression of the disease after a mean observation of 5.5 years.
The clinicopathologic findings are very similar in both adults and children, including an indolent clinical course: multiple papules, plaques and/or nodules present on the arms, upper trunk, and face. Most recently, a 15-year-old adolescent boy with multiple lesions representing PCMZL and juxta-articular fibrotic nodules showing a pattern of nodular sclerosis with peripheral plasma cell-rich infiltrates has been reported.
236
Histologically, early lesions have a perivascular and periadnexal infiltrate, while more advanced ones may have a nodular and/or diffuse cellular infiltrate extending into the subcutaneous fat. There are small lymphocytes, lymphoplasmacytic cells, and plasma cells that demonstrate monotypic cytoplasmic Ig on routine histology. Reactive T-cells may be present and can represent a significant proportion of the infiltrate. It is not uncommon to see a varying component of plasma cells, eosinophils, and histiocytes. In MZL, reactive but small and atrophic lymphoid follicles are often seen (Figure 11).230,237
Cutaneous marginal zone lymphoma (MZL). There is a dermal-based infiltrate with adnexotropism and sparing of the surface epidermis. The lymphoma cells are medium in size and have abundant cytoplasm with a monocytoid appearance.
By IHC, marginal zone cells show expression of CD20, CD79a, and BCL-2 and are usually negative for CD10, CD5, and BCL-6 (Figure 12). Coexpression of CD43 is rare in the cutaneous sites. Tumors with prominent plasma cell differentiation show expression of CD138. IgG4, while found commonly in lesions from adults, has not been seen in the pediatric group.229,238 Clusters of plasmacytoid dendritic cells, using a CD123 immunostain, can be seen. Only one of the cases in a child has been found in association to B. burgdorferi. In situ hybridization for kappa and lambda and/or immunohistochemical stains for surface light chains can be useful in detecting a restricted population of plasma cells. Clonality studies for IGH gene rearrangements are also helpful to prove a clonal population of B-cells.
239
Similarly to adults, trisomy 3 can be seen using FISH where it is the most frequent chromosomal aberration seen in this type of lymphoma in adults, with approximately 20% of studied cases displaying this anomaly, either singly or in combination with t(14;18)(q32;q21) or t(3;14)(p14.1;q32).240–242 The t(11;18) translocation, linked to gastric and lung MALTs, has not been seen in cutaneous sites. Guitart and Gerami
243
have proposed the term atypical marginal zone hyperplasia to describe a subset of cases in young individuals with lambda-restriction and lack of BCL-2 expression. This term has been previously applied by Attygalle et al.
244
to subsets of cases presenting in the tonsils of young children which are polyclonal by gene rearrangement studies and with CD43 coexpression by IHC. However, a word of caution should be taken, with the use of atypical marginal zone hyperplasia, as some consider those as bona fide cases of MZL.
239
Swerdlow et al.
245
have also argued that many examples of “cutaneous lymphoid hyperplasias” with the presence of germinal centers are actually examples of CMZL.
Cutaneous marginal zone lymphoma (MZL)—IHC. The lymphoma cells are positive for CD20, while negative for BCL-6. D2-40 highlights focal dendritic cell networks. In situ hybridization for kappa and lambda show kappa restriction. Ki67 shows a very low (<10%) proliferative index.
The differential diagnosis of PCMZL in children includes Borrelia-associated lymphocytoma cutis, which mainly affects the ear lobes, nipples, and scrotum in children and presents with a solitary lesion in most patients, which contrasts to the multifocal lesions in the majority of PCMZL. 246 The so-called tibial lymphoplasmacytic plaque occurs in the latter location. 247 Some regard this lesion within the spectrum of linear acral pseudolymphomatous angiokeratoma of children. Cutaneous plasmacytosis and plasmacytoma are now regarded as MZL. Among other B-cell lymphomas, primary cutaneous follicle center lymphoma (PCFCL) has a germinal center phenotype (BCL-6 and ± CD10). A prominent plasmacytic infiltrates can be seen in Melkersson–Rosenthal syndrome, usually in association with granulomas with an intralymphatic distribution.
Primary Cutaneous Follicle Center Lymphoma
PCFCL is an indolent neoplastic proliferation of B-cells with a germinal center phenotype. 25 It is one of the most common forms of cutaneous B-cell lymphomas in middle age and older individuals (median: 51–58 years), but extraordinarily rare in children. 248 By definition, PCFCL is limited to the skin at the time of diagnosis, without evidence of systemic or nodal involvement, and is a distinct entity separate from systemic or nodal follicular lymphoma (FL). It typically presents as a solitary or clustered smooth, erythematous to violaceous infiltrative papules, plaques, nodules, or tumors, in the head and neck or upper trunk. 249 Armitay-Laish et al. 234 reported the occurrence of a spindle cell variant of PCFCL in a 17-year-old adolescent girl. Condarco et al. 250 reported another case of PCFCL in the scalp of an 8-year-old boy. In the large series (n = 69) of pediatric cutaneous NHL from Gratz, a single case of a PCFCL in a 20-year-old individual has been reported. 3 Another case of PCFCL in the setting of the rare congenital immunodeficiency WHIM (warts, hypogammaglobulinemia, infections, and myelokathesis) syndrome has been described. 251 Another case was reported in a 16-year-old adolescent boy of PCFCL arising in the nose and extending into the maxillary sinus and soft palate. 252
Histologically, PCFCL has a nodular, nodular and diffuse, or diffuse growth patterns in the dermis with sparing of the surface epidermis (a Grenz zone separating the infiltrate from the epidermis is invariably present).
253
The infiltrate can extend into the adipose tissue. As opposed to systemic FL, grading has no prognostic significance as all cases of PCFCL show an indolent course. The areas with abnormal follicles lack the tingible body macrophages present in reactive germinal centers (Figure 13). The neoplastic cells are composed of small cleaved centrocytes and larger centroblasts.254–256 The lesions with a diffuse growth pattern show a predominance of centroblasts and can be potential mimickers for large B-cell lymphomas. There is a variant composed largely of spindle cells (sarcomatoid form), originally referred as “reticulohistiocytoma dorsi” or “Crosti’s lymphoma.”257,258 In such, fascicles of spindle cells admixed with areas of atypical centroblasts and centrocytes are present. One of the reported pediatric cases showed the spindle cell cytology. By IHC, PCFCL is positive for CD19, PAX-5, CD20, CD79a, and BCL-6. CD10 is variably positive, depending on the pattern of growth (nodular growth is more typically positive, whereas diffuse growth is negative). BCL-2 is positive in approximately 50% of cases. The follicular dendritic networks can be highlighted by a CD21, CD23, and D2-40 immunostains. Clonality studies can be used to demonstrate a rearrangement of the IGH gene. In situ hybridization can also show on occasion a clonal population of B-cells. Translocations of the IGH-BCL2 genes t(14;18) can be present in 18% to 41% of PCFCL.259,260
Primary cutaneous follicle center lymphoma—nodular pattern. There is a dense infiltrate in the dermis with sparing of the epidermis. The infiltrate shows a nodular appearance. The poorly formed follicles lack tingible body macrophages or polarization. In this particular case, many centroblasts are present, which can potentially raise the consideration of a large B-cell lymphoma. The infiltrate is positive for CD20 and BCL-6, while is negative for BCL-2. This is one of the most characteristic phenotypes of PCFCL.
Differential diagnostic considerations with PCFCL include reactive lymphoid hyperplasias, PCMZL, cutaneous involvement by systemic FL, and diffuse large B-cell lymphomas (DLBCL, particularly the so-called leg-type lymphoma). PCMZL differs in the lack of germinal center markers. Although PCMZL can show atrophic germinal centers, those typically have a very high proliferation and are rather small, in counterpart with the neoplastic follicles of PCFCL that have a lower Ki67. In situ hybridization can help highlight a clonal population of plasma cells. Some cases of PCZML can also show a high proportion of IgG4+ plasma cells. Cutaneous involvement by systemic FL has never been documented in the pediatric setting as systemic FL is a disease of older individuals and systemic pediatric FL is a more localized disease. DLBCL leg type has not been documented in the pediatric age and is a disease of older adults. In counterpart, DLBCL-LT presents clinically in the legs and has a nongerminal center profile (MUM1 strongly positive).
Precursor T/B-cell Lymphoblastic Leukemias/Lymphomas
Acute lymphoblastic leukemia/lymphoma (ALL) is the most common type of leukemia in childhood, representing 80% of cases. 261 In the United States, the incidence of ALL is about 30 cases per million persons younger than 20 years of age. The peak incidence occurs between the ages of 3 and 5 years. There is a slightly higher incidence of ALL in boys than girls. 262 Approximately 80% to 85% of cases are B-ALL and 10% to 15% T-ALL. 263 Although the etiology of the disease is still largely unknown, certain genetic disorders have a greater predisposition for the development of ALL, including trisomy 21 and ataxia-telangiectasia. Some cases are associated with MLL gene rearrangements and might be secondary to chemotherapy.
Between 5% and 20% to 30% of patients develop cutaneous dissemination. Nearly 50 to 60 cases of B-ALL have been reported in children with skin involvement.4,263–265 The frequency of cutaneous involvement in T-ALL is less well documented. However, Lee et al. 266 reported an incidence of 4.3% in T-ALL and 15% in B-ALL. Approximately 33% of patients with the lymphomatous variants had cutaneous involvement, and only 1% in those with leukemic presentations. The lesions present as erythematous or violaceous nodules, tumors, or plaques (sometimes can be multiple). The distribution of the lesions is typically the head and neck, upper trunk, and abdomen. In a series of 6 cases, 264 most presented between the ages of 5 and 15 years, and all occurred in girls. Occasional cases of aleukemic forms have been reported.263,267–269 A single case with a congenital presentation has also been described in association with an MLL rearrangement. 270 Cases of T-ALL are frequently associated with a mediastinal mass (50%–65%). Many cases of ALL present in neonates with multiple blue/purple plaques or nodules on the skin, hence the term “blueberry muffin syndrome.”
The histopathologic findings for both B and T-ALL are identical.265,271 There is a diffuse and dense dermal infiltrate with extension into the subcutaneous tissue (Figure 14).4,234,263,264,268–270,272 A Grenz zone is typically present with epidermal sparing. Cytologically, the infiltrate consists of small lymphoblasts with scant cytoplasm, fine nuclear chromatin, irregular nuclei, and inconspicuous nucleoli. Linear arrangements of lymphoblasts forming files can be noted. Infiltration of the skin adnexae is also frequently seen as well as nerves and piloerector muscles. Mitotic activity is typically brisk.
B lymphoblastic leukemia/lymphoma with skin involvement. There is a diffuse involvement in the dermis by a mononuclear cell infiltrate. The blasts are medium in size and have finely dispersed chromatin and variable nucleoli. The blasts are positive for CD34 and CD10. In this particular case, the child’s ALL also had the t(9;22) translocation.
By IHC, the blasts of B-ALL express CD19, PAX5, CD79a, and CD43. TdT and CD34 are often expressed in the malignant cells, and CD99 can be seen in >50% of cases. Expression of CD45 is dim or negative, and most cases are negative for CD20, but CD10 is present in most, but not all cases. B-ALL with MLL rearrangement is typically CD15+ and CD10−. Those cases of B-ALL with the t(12;21) translocation (Philadelphia chromosome) have a worse prognosis and are associated with CD10 coexpression.
The immunophenotype of cells in T-ALL usually includes positivity for TdT with variable expression of CD45, CD34, CD1a, CD10, CD2, cCD3 (lineage specific), CD4, CD5, CD7, and CD8. CD4/CD8 coexpression is most frequently observed; however, coexpression is not necessarily a marker of an immature phenotype because cases of mature T-cell lymphomas have been described with dual positivity. More reliable markers to confirm an immature phenotype include TdT, CD1a, CD99, and CD34. 273 According to the expression of specific markers T-ALL can be subdivided into early or pro-T, pre-T, cortical-T, and medullary-T. The molecular background of T-ALL is less well understood. Cytogenetic abnormalities are frequent in T-ALL cases (50–70%). The most common cytogenetic abnormalities involve 14q11-13 the site of TCR alpha/delta, including inv(14)(q11;q32) and deletions or translocations involving chromosomes 9, 10, and 11 corresponding to sites of TCR alpha, beta, and gamma subunit genes found in 47% of T-ALL. Some cases can also present in association with myeloid hyperplasia, eosinophilia, and the t(8;13)(p11;q11) aberration affecting the FGFR1 gene. The latter has now been referred to as the 8p11 myeloproliferative disorder.274,275 Similarly to B-ALL, T-ALL is typically associated with rearrangements of the TCR.
The differential diagnosis can be challenging in the primary lymphomatous lesions or its aleukemic forms. The differential diagnosis in “blueberry muffin” syndrome includes neuroblastoma, myeloid leukemia, rhabdoid tumor, and rhabdomyosarcoma. In fact, a case originally diagnosed as Ewing sarcoma (EWS) represented an example of B-ALL with CD99 coexpression. 268 However, EWS does not express B-cell antigens. Indeed, CD99, CD34, and TdT in B-ALL are useful in the differentiation from mature B-cell lymphomas in the skin, which overall, are extraordinarily uncommon. In the differential diagnosis, Merkel cell carcinoma may rarely present in children276,277 and show CD99 expression similarly to B-ALL. CK20 and NSE can help in the distinction between the two. Cutaneous involvement by neuroblastoma is also rare and might be a pitfall, but CD99 is negative in the latter.278,279 However, CD99 expression is more typically cytoplasmic, rather than the crisp staining seen in EWS. 280 Neuroblastomas are also positive for neuroendocrine markers. Other small round cell sarcomas can be distinguished with the presence of specific markers (myogenin, desmin, etc).
Cutaneous Myeloid Neoplasms
Leukemia cutis (LC) is manifested by a cutaneous infiltration of neoplastic myeloid cells, lymphoid blasts, or mature lymphoma cells. AML, particularly in those with monocytic or myelomonocytic differentiation occur as LC. 281 However, LC is seen in association with chronic myelogenous leukemia, myelodysplastic syndromes, and acute lymphoblastic leukemias (T-ALL and B-ALL).282–285 Myeloid sarcoma, extramedullary myeloid tumor, granulocytic sarcoma, and monocytic sarcoma are often termed LC when occurring in the skin, but many now prefer the term “myeloid sarcoma” when there is a nodular infiltrate of immature blasts in the dermis. 286 In contrast, LC is the preferred term when the malignant infiltrate shows a diffuse, interstitial pattern through the dermis with the clinical appearance of a rash. LC in children may present at or soon after birth.
Rarely, cutaneous involvement by a leukemic infiltrate can occur in the absence of bone marrow or peripheral blood involvement by acute leukemia; this then is referred to as aleukemic leukemia cutis (ALC)267,287–295 or aleukemic myeloid sarcoma.281,296 Byrd et al. 286 proposed using aleukemic to describe those cases of extramedullary involvement in the absence of blood and bone marrow disease for at least 1 month. The literature also includes cases described as aleukemic despite concomitant bone marrow disease in patients who do not have circulating peripheral blasts.297,298 An adverse prognosis has been associated with, while others have proposed that ALC is a heterogeneous condition that can even result in spontaneous resolution without the need for aggressive chemotherapy.235,286,299–301 We have recently published a series of cases of ALC including some with spontaneous resolution, and in 2 cases, there was the presence of MLL rearrangements. 302 ALC can also be seen in association with pre–B-cell ALL267,294,297 and in rare cases of T-cell ALL.269,303 A monocytic phenotype and certain cytogenetic aberrations, such as t(8;21) or inv(16), are found in a disproportionate number of cases. In addition, the expression of T-cell markers and CD56 is more frequent in extramedullary myeloid blast cells.286,304 Otsubo et al. 305 reported the development of an acute promyelocytic leukemia following ALC in association with NPM-RARA fusion transcript in a child. Agrawal et al. 299 reported ALC in siblings at birth. Interestingly, 11q23 abnormalities have been detected by FISH in some cases of congenital LC, which strongly raises the likelihood of abnormalities in the MLL gene.306,307 Torrelo et al. 308 described a very aggressive clinical presentation of ALC in a newborn, clinically manifested as “blueberry muffin” skin lesions. Transient abnormal myelopoiesis (TAM) is an entity characterized by the presence of blasts in the blood of children with trisomy 21, which in many cases can be reversible. Cutaneous lesions with blast infiltration have been reported in children with TAM.309–311
Cutaneous interstitial deposits of immature blasts present as papules, nodules, or plaques and are found in 2% to 12% of AML cases. Although less common, LC occurs in the setting of lymphoblastic leukemias, with an estimated incidence of 1% to 3%.
312
The diagnosis of ALC is a challenging one not only because of its infrequency but also due to the absence of concomitant blood or bone marrow involvement. The diagnosis can also be elusive because the infiltrate can adopt a very sparse perivascular and periadnexal distribution.
313
Morphologically, the leukemic cells are centered in the dermis and may extend into the subcutaneous tissue (Figure 15).
314
Epidermotropism is usually not seen. The malignant cells can disrupt the vessels and adnexal structures, and a “leukemic vasculitis” can also occur.
315
The vasculitis is thought to represent direct endothelial injury by the leukemic cells.
316
Leukemia cutis (LC). Leukemic blasts show diffuse infiltration in the dermis. The blasts are small to medium in size. A subset of the blasts is positive for CD34 and MPO. All the leukemic cells are CD43+.
A diagnosis of LC is invariably corroborated by IHC. Because of the frequent monocytic lineage, CD34, TdT and CD117 are often not helpful in identifying immature blasts, but Cibull et al. 314 found that CD68 and lysozyme were additionally helpful in the diagnosis. The CD68 KP1 clone appears to be more specific and reliable to identify the myeloid origin of the cells. 317 On the other hand, Cronin et al. 318 suggested that the combination of CD43 and CD68 was more reliable. Amador-Ortiz et al. 319 with a panel including CD117, CD33, and lysozyme confirmed most cases of cutaneous myeloid sarcoma. The addition of CD14 and KLF-4 were also useful in achieving a greater degree of sensitivity and specificity in those lesions of monocytic lineage. ERG is a more recent marker that has been used with success to help in the diagnosis of LC. 320 However, a problem with the use of immunostains is that numerous histiocytic disorders (benign and malignant) in cutaneous sites have a similar phenotype. In fact, some LCs have been masked by the presence of a prominent granulomatous reaction in the dermis, as reported by Tomasini et al. 298 Ideally, correlation with the bone marrow immunophenotypic findings of the immature blasts could be helpful. Interestingly, discrepancies in the phenotype of the immature cells in the skin and the bone marrow have been documented, a feature that can complicate the diagnosis further.318,319 Other molecular tools can sometimes be helpful in establishing a diagnosis, such as MLL rearrangements by FISH or NPM1 and FLT3-ITD mutations by PCR analysis.
Blastic Plasmacytoid Dendritic Cell Neoplasm
BPDCN is an extremely rare subtype of acute leukemia and was formerly known as blastic natural killer (NK)-cell lymphoma or CD4+/CD56+ hematodermic neoplasm.321–324 Once postulated to originate from NK-lineage precursors, accumulating phenotypic, functional, and genetic evidence has pointed to its derivation from hematopoietic precursors with commitment to the plasmacytoid dendritic cell lineage cells which are positive for CD123.323,325–329 BPDCN is rare in children,224,321,330–333 and the largest series from BPDCN in the pediatric setting was reported by Jegalian et al. 225
Clinically, most cases have cutaneous lesions at diagnosis in adults (85%), but the opposite has been found in children: 7 of the 29 cases (24%) lacked cutaneous disease at presentation, with disease confined to the bone marrow, peripheral blood, lymph nodes, spleen, and/or liver. 225 Those without cutaneous disease at presentation had 100% survival at 60 months’ follow-up. Most adults relapsed within the first year of diagnosis with bone marrow and central nervous system (CNS) involvement (approximately 33% of cases). The disease has an overall survival of approximately 74% in children. 225
CD123, CD4, and CD56 positivity, the most common BPDCN immunophenotypic profile, is not specific since these markers are also expressed in both AML and ALL.
332
However, cases without cutaneous involvement also expressed other BPDCN markers, including BDCA-4, CD303/BDCA-2, CD2AP, and TCL1. CD68 (KP1) is generally negative, but focal punctate staining is seen in a minority of cases. Strong staining for CD68 should raise the suspicion of acute or chronic myeloid leukemia with monocytic differentiation.
225
Myeloid cell nuclear differentiation antigen can be used effectively to distinguish between LC and BPDCN, since only LC is positive.
334
Morphologically, BPDCN in children tends to have smaller sized cells and a lymphoblast-like appearance of the blasts. In adults, BPDCN has a more pleomorphic appearance (Figure 16). In addition, S100 expression is seen in approximately 75% of pediatric cases, compared to only 25% of cases in the adult population.
335
The genetics of this disease is poorly understood, but recently, one case was reported with EWSR gene rearrangement, an important diagnostic consideration in the pediatric population.
336
TET2 is the most common mutated gene (36%–80%)
337
; other recurrent somatic mutations include IKZF3 and ZEB2 (12%–16%),
337
ASXL1 (32%), NPM1 (10%), and RAS (9%–27%).
338
Gene expression profiling has demonstrated a unique signature, distinct from AML and ALL.
339
Compared with normal plasmacytoid dendritic cells, BPDCN has shown increased expression of genes involved in Notch signaling and NFKB activation.
Blastic plasmacytoid dendritic cell neoplasm (BPDCN). A dermal-based proliferation of monocytoid cells is noted with epidermal sparing. The cells show a very blastic appearance. The malignant cells are positive for CD56 and TdT. Careful recognition is imperative so such process is not confused with an acute leukemia.
Cutaneous Histiocytosis in Children
A more recent update in the classification of histiocytic disorders of adults and children has been proposed, which provides integration of cell of origin of these disorders, in addition to common molecular pathways that appear to be altered in this group of neoplasms. To this extent, histiocytoses can be divided into 5 major categories: (1) Langerhans-related (“L”), (2) cutaneous and mucocutaneous (“C”), (3) malignant histiocytoses (“M”), (4) Rosai–Dorfman disease (RDD; “R”), and (5) HLH and macrophage activation syndrome (“H”). 340 The “L” group includes Langerhans cell histiocytosis (LCH), indeterminate cell histiocytosis, Erdheim–Chester disease (ECD), mixed LCH/ECD, and the disseminated forms of juvenile xanthogranuloma (JXG). They all share clonal mutations involving genes in the mitogen-activated protein kinase (MAPK) pathway in more than 80% of cases; the “C” group incorporates the cutaneous xanthogranulomas (JXG, adult xanthogranuloma, solitary reticulohistiocytoma, benign cephalic histiocytosis, generalized eruptive histiocytosis, and progressive nodular histiocytosis [PNH]) and nonxanthogranulomatous disorders (cutaneous RDD and necrobiotic xanthogranuloma). The “R” group includes familial RDD, in addition to the sporadic extracutaneous forms (classical, extranodal, etc). The “M” group is an extraordinarily rare group of disorders that include histiocytic, interdigitating dendritic cell, Langerhans cell, or indeterminate cell sarcomas. Finally, the “H” group includes the primary and secondary HLH. In this article, we will only focus on the diseases that primarily affect the skin.
Langerhans Cell Histiocytosis
LCH is a rare clonal histiocytic proliferation without any known predisposing risk factors, although linkage to inflammation, cytokine release, and immune dysregulation has been proposed over the years. Langerhans cell (LC) proliferations include reactive LC hyperplasia, LCH, Langerhans cell sarcoma, and congenital self-healing reticulohistiocytosis.341,342 Debate still lingers on the nature of LCH, almost 20 years after the publication of the study with the demonstration of clonality in nonpulmonary LCH lesions. 343 Newer data now show evidence of a clonal myeloid cell origin for LCH.344–349 Despite a rare overall incidence in the general population, LCH has a strong predilection for children and particularly an aggressive clinical course in the very young individuals, typically less than 2 years of age at diagnosis.343,350,351
LCH can involve any site or organ system and is unifocal, multifocal, or systemic in nature. Common sites of involvement include skin, bones (especially the skull), CNS, lungs, liver, and spleen. This variability in distribution and extent of involvement best explains the long list of clinical designations under the LCH umbrella. 352 The cutaneous lesions, much like the overall clinical spectrum of LCH, are variable in appearance: cutaneous involvement is most common in multisystem disease in infants and children. In multisystem disease, the skin lesions are erythematous and maculopapular or nodular with ulceration at times. These lesions often involve the scalp, intertriginous areas, including the perineal and perianal sites and the trunk.353,354 The skin lesions may resemble seborrheic dermatitis or eczema; crusting, scaling and alopecia often accompany the scalp lesions. Erosive intertrigo of the axilla, perianal region, and other flexural folds is well-described and may mimic diaper rash in infants. 355 Isolated skin involvement by LCH is infrequent and occurs in less than 5% of all cases. Skin-limited LCH occurs in the form of one or multiple, often asymptomatic but occasionally erosive, papules or nodules with surrounding erythema or rarely forming masses with or without ulceration. One study documented mucosal involvement—most frequently the oral or genital mucosa—in 20% of cases with otherwise isolated skin LCH. 353
Systemic symptoms including fever, lymphadenopathy, hepatosplenomegaly, and cytopenias can be seen in multisystem disease, together with clinical manifestations of organ dysfunction such as diabetes insipidus in cases with pituitary involvement. Such clinical manifestations warrant the staging of LCH using bone scans and magnetic resonance imaging of the brain, in addition to blood evaluation for electrolyte imbalances.
A common unifying theme in all of these lesions/clinical entities is the histologic demonstration of LCs in aggregates despite the admixture with eosinophils, macrophages, lymphocytes, neutrophils, and nonneoplastic multinucleated giant cells (Figure 17).356–358 The diagnosis of LCH can be tentatively made based on the characteristic morphologic findings of the LCs which are medium to large in size, exhibit abundant eosinophilic cytoplasm, and have a “coffee bean” -shaped grooved nuclei. These characteristic features although best seen on cytologic preparations can also be appreciated in a well-fixed and prepared histologic section. Involvement of the skin may be confined to small collections of cells at the dermo-epidermal interface with variable basal vacuolar degeneration or as dense infiltrates throughout the dermis with or without the formation of eosinophilic abscesses. Marked epidermotropism is also frequently seen. Confirmation of the diagnosis is achieved by demonstrating Langerhans granules (Birbeck granules) on electron microscopy
359
or—more easily—by the characteristic positivity of LCs for CD1a, S100, and langerin. The langerin immunostain is used, by many, as a surrogate marker for the presence of the Birbeck granules.360–364 A careful distinction between reactive dermatoses with associated Langerhans cell hyperplasias and LCH is important to be made, in order to avoid overdiagnosing LCH. LCH typically fills the papillary and/or deeper dermis and sometimes can show a perifollicular and epidermal distribution. Chronic dermatoses often show increased number of perivascular and perifollicular dendritic cells that are CD1a+/langerin low to negative. Contact dermatitis shows frequent small intraepidermal clusters of Langerhans cells that lack significant cytologic atypia.
365
Langerhans cell histiocytosis. A perifollicular and dense dermal histiocytic infiltrate is noted. The cells have eccentric nuclei, with a reniform appearance and the presence of nuclear grooves. The Langerhans cells are positive for CD1a and langerin.
In neonates and young infants, when involvement is limited to skin, a variant of LCH known as congenital self-healing LCH (CSH-LCH or Hashimoto–Pritzker disease) should be separated based on clinical histologic and ultrastructure grounds.366–368 In CSH-LCH, lesions typically involve the deep dermis in a nodular fashion, unlike conventional LCH lesions where lesions are mainly in upper dermis, extending to epidermis. Both clinical entities show similar staining with CD1a, S100, and langerin. Langerhans cells of CSH-LCH may show Birbeck granules similar to conventional LCH cells by electron microscopy; however, they can be separated ultrastructurally by the presence of unique cytoplasmic dense bodies containing concentrically arranged laminar structures. It is important to emphasize that CSH-LCH should be evaluated appropriately and followed closely, as some cases can persist and progress to LCH.369,370
Although the initial report of clonality in LCH has not been followed by a demonstration of any gross chromosomal abnormalities in 72 cases of LCH, more recently, BRAF V600E mutations have been identified in approximately 50% of cases with an association with younger age patients but not with the site or stage of disease. Berres et al. 347 found a prevalence of 64% for BRAF mutations in LCH and particularly a high-risk of recurrence among the mutated cases. Heritier et al. 371 found that the mutation showed correlation with high-risk features, increased risk of recurrence, and increased resistance to first line therapies. Whole exome sequencing subsequently identified the presence of MAP2K1 (MEK1) mutations in 7 of 21 cases of LCH with wild-type BRAF and in none of 20 BRAF V600E mutant cases. These studies implicate aberrations of the MAPK signaling pathway in the pathogenesis of LCH and strongly support a neoplastic pathogenesis.372–375
Treatment protocols of LCH take into consideration sites of involvement and extent of disease. Patients’ age is an important prognostic indicator (an interesting phenomenon observed also in certain pediatric tumors such as neuroblastoma). Contrary to the latter tumor, presentation in infancy is an unfavorable prognostic factor since there is often bone marrow, liver, and spleen involvement. 376
Erdheim–Chester Disease
First described in 1930 by Jakob Erdheim and William Chester and named in 1972 by Jaffe, ECD is a rare disseminated histiocytosis that is uncommon in children377,378 and typically presents in adults over 50 years of age, more commonly in men. Since approximately 50% or more of cases of ECD have a BRAF V600E mutation, this entity is now included under the category of Langerhans cell (LC)-related histiocytosis (Group L). 340 In addition, mutations in the MAPK (such as in NRAS) and PIK3 pathways have been reported. Another finding in support of a LC relationship is the fact that 20% of biopsies of ECD have coexistent LCH.340,379 However, these data were only based on small case series and case reports. It is predominantly a visceral disease and any organ system can be involved, notably the bones, heart, lungs, retroperitoneum, and CNS. Bilateral, symmetric sclerosing bone lesions in the metaphysis of long bones, usually the distal femur and proximal tibia and fibula, are the diagnostic imaging features.349,374,375,377,380 It should be emphasized that ECD is a clinicopathologic diagnosis that requires the presence of the clinical, radiographic, and histopathologic findings.
The skin is involved in about 25% to 30% of cases; these patients most often develop red-yellow xanthoma-like papules on the extremities, trunk, axillae, eyelids, and groin. The lesions may coalesce into plaques with time. Pigmented lesions may also been seen on the oral mucosa. An infiltrate of foamy, xanthomatous histiocytes includes the dermis and may extend into the subcutaneous tissue. Dense fibrosis or sclerosis may be present as well. The histologic features are very similar to JXG with their xanthomatous features and even Touton giant cells. 381 In children, ECD can occur in association with lymphoblastic leukemias and LCH.377,378,382,383 ECD can also have a presentation that mimics multicentric reticulohistiocytosis. 384
The clinical differential diagnosis includes other systemic diseases, such as systemic JXG, xanthoma disseminatum (XD), RDD, LCH, sarcoidosis, amyloidosis, and storage disorders. The histiocytic cells of ECD are positive for CD163 and CD68, while negative for CD1a and langerin. Some cases can have partial expression of S100. In addition, a recent study has shown expression of the Programmed Cell Death 1 (PD-L1) protein in a subset of histiocytoses, including ECD disease, suggesting a possible role of immune checkpoint blockade in treatment. Rare cases with histologic and immunophenotypic features of ECD, but with strong ALK expression and rearrangement of the ALK gene, can be seen. Such cases are examples of ALK+ histiocytosis of childhood and not ECD. 385 ECD is often progressive and fatal despite aggressive therapy. The recent identification of various mutations in ECD has resulted in some success with targeted therapies, such as with vemurafenib. In a recent pangenomic analysis, all cases of ECD had at least one mutation activating the MAPK pathway. 373 In addition, in-frame fusions involving several kinases including BRAF, ALK, and NTRK1 were identified in ECD without BRAF, MAP2K1, or N/KRAS point mutations.349,374,375 ECD should also be distinguished from JXG. As opposed to JXG, ECD more frequently harbors the BRAF V600E mutation. ECD also has the typical clinical and radiographic findings, not present in cases of JXG. The recently reported cases of BRAF mutated JXG are now believed to be examples of ECD.386,387
Juvenile Xanthogranuloma
JXG is the most common form of non-LCH and is currently classified as group “C” for the cutaneous limited forms, and in the “L” category for the systemic variants. 340 The exact incidence is unknown as most cases are diagnosed based upon clinical findings and without histologic confirmation. One study reported that JXG represents only 0.5% of pediatric tumors. Most cases are diagnosed in infants and young children, most often in the first year or second year of life.388–390 Males are slightly affected more often than females, especially when lesions are multiple. Less commonly, JXG occurs in adolescents and young adults and in some cases may be difficult to differentiate from ECD, except for the BRAF V600E mutations.
A solitary, well-demarcated, firm, round to oval dermal nodule, typically located in the head and neck region, but also seen on the trunk or upper limbs is the clinical presentation in 70% to 75% of cases. Less common cutaneous sites include the genitalia, soles, and fingers.272,380,391,392 The shape of the lesion can vary greatly, as it may be keratotic, pedunculated, linear, flat, or plaque-like. The papules or nodules are usually 5 to 10 mm in diameter but can measure up to a few centimeters.393–395 Initially, the lesion is raised and pink to erythematous in color, but over time, becomes yellow-brown and may flatten as the histiocytes undergo xanthomatous transformation and regression. Regression may occur over a period of months to years with residual hyperpigmentation, atrophy, or anetoderma. Telangiectatic blood vessels or ulceration may be seen overlying the lesion which imparts a resemblance to pyogenic granuloma. The presence of multiple skin lesions occurs infrequently, in less than 10% of cases. 388 Although JXG is typically limited to the skin in the majority of cases, it may arise in extracutaneous sites with or without skin involvement. The most common extracutaneous site is the subcutaneous or deep soft tissue, which occurs in about 15% of cases. Multifocal visceral and osseous sites are involved in about 5% of cases and may include the oropharynx, nasopharynx, gastrointestinal tract, liver, lung, testis, spleen, lymph nodes, CNS, and kidney. Most systemic cases occur in young children under the age of 2 years. Ocular JXG occurs in less than 1% of children with multiple JXGs, typically in young children, and presents without cutaneous involvement in about 60% of cases.
JXG can be associated with neurofibromatosis type 1 (NF1) and juvenile myelomonocytic leukemia (JMML). Nearly 5% to 10% of NF1 patients and up to 30% of NF1 patients less than 2 years of age have been reported to have JXG. The presence of cafe au lait macules and JXG should suggest the likelihood of NF1. The association among JXG, NF1, and JMML, while well described, is poorly understood in terms of the pathogenetic relationships. JXG usually precedes JMML; however, it is unclear if JXG is a reliable marker for the subsequent development of JMML in NF1 patients. It has been suggested that mutations in the GTPase neurofibromin, which is present in NF1, leads to RAS dysfunction and downregulation of Fas antigen with subsequent dysregulation of apoptosis in hematopoietic cells with the development of leukemia and JXG. Some cases of JXG follow the diagnosis of LCH.272,396–402 More recently, systemic presentations of JXG with CNS involvement have shown BRAF V600E mutations.386,387 The question that arises from these is: Should those cases be qualified as ECD/L group lesions that have not presented themselves with systemic findings (ie, brain only), or is there a truly separate group of JXG/BRAF V600E positive tumors?
A dense mononuclear histiocytic infiltrate in the dermis extending to the interface with the epidermis is present (Figure 18).388,403,404 In smaller lesions, the infiltrate may be limited to the superficial dermis but, in larger lesions, may extend into the subcutis. There is often flattening of the epidermis with loss of the rete ridges; however, the epidermis and adnexa are spared. The infiltrate is composed of small mononuclear cells as well as multinucleated giant cells with and without the features of Touton giant cells. In fact, cutaneous JXG may be devoid of giant cells and their presence in extracutaneous lesion is often difficult to demonstrate. Lymphocytes, eosinophils, plasma cells, neutrophils, and mast cells may be scattered throughout the infiltrate or altogether absent. The morphology of the histiocytes can vary depending on the “age” of the lesion which is often difficult to determine with any certainty. The so-called early lesions are composed predominantly of mononuclear cells with abundant eosinophilic and minimally vacuolated cytoplasm. More “mature” lesions contain histiocytes with “lipidized or xanthomatized” cytoplasm with numerous fine cytoplasmic vacuoles. Touton giant cells, which are characterized by a ring or wreath of nuclei surrounded by a rim of foamy cytoplasm, are a characteristic, but their presence is variable in this phase. Scalloped histiocytes with an angulated or jagged border and spindled histiocytes may also be seen. In fact, some lesions are composed predominantly of spindle cells, but the finely vacuolated interspersed mononuclear cells are the clue to the diagnosis of JXG. Not entirely surprising is the resemblance in some cases between JXG and dermatofibroma (DF). Mitotic figures may be seen but atypical mitotic figures are absent.
Juvenile xanthogranuloma (JXG). A dermal and nodular histiocytic infiltrate is present. Many xanthomatized histiocytes (lipidized cells) and Touton-type giant cells are seen. Courtesy of Path Presenter.
The histiocytes stains were positive for CD68, CD163, CD4, CD14, factor XIIIa, HLA-DR, fascin, vimentin, and lysozyme and negative for CD1a and langerin.388,404 The histiocytes are usually negative for S100, but it is not unusual to encounter individual and patchy staining in some cases. Frequent admixed S100 dendritic cells can be seen in some cases. Ultrastructural examination of “mature” lesions shows lipid vacuoles, lysosomes, cholesterol clefts, comma-shaped bodies, and myeloid bodies in the cytoplasm of the histiocytes. No Birbeck granules are present.
The differential diagnosis of JXG is often with LCH. As opposed to JXG, LCH more typically infiltrates the epidermis and only rarely involves the reticular dermis. In addition, the LC shows reniform nuclei and lacks the lipidized changes and Touton cells. Immunohistochemical stains are often needed in order to distinguish with some certainty between these entities. In contrast to the histiocytes of JXG, LCs cells are positive for S100, CD1a, and langerin. The lipidized variant of DF contains numerous large, foamy cells and Touton-like giant cells, reminiscent of JXG. However, areas of spindle cells in a whorled or storiform pattern may be present and collagen trapping can be seen at the periphery of the lesion. However, the distinction between JXG and DF in an older child is a challenge with some degree of uncertainty. DFs tend to have overlying epidermal changes, such as acanthosis, hyperpigmentation of epidermal basal cells, or folliculosebaceous induction, and often contain hemosiderin unlike JXG. Both lesions can be positive for factor XIIIa, but DFs are usually negative or only weakly positive for CD68.
Reticulohistiocytoma can also mimic JXG clinically and histologically (Figure 19). The histopathologic findings of reticulohistiocytoma, including the presence of densely eosinophilic “ground glass” cytoplasm, usually allow for a distinction from JXG; however, there may be immunohistochemical overlap between these lesions as the reticulohistiocytomas are also positive for CD68 and CD163, negative for CD1a, and often negative for S100. It should be emphasized that some consider reticulohistiocytomas as histologic variants of JXG.340,365,405 Spitz nevi can also enter the differential diagnosis; however, the melanocytic nature of the lesion can readily be demonstrated with the application of the melanocytic markers, Melan-A, HMB-45, or SOX-10. Mast cell neoplasms (mastocytomas) can also resemble JXG (when the latter are devoid of lipidized cells). Mastocytomas are characteristically positive for CD117, tryptase, and have aberrant coexpression of CD2 and CD25. Alpha chloracetate esterase (Leder stain) is positive in the mast cell granules.
Reticulohistiocytoma. Dermal-based histiocytic infiltrate with an epithelioid appearance. The histiocytes are large and show markedly eosinophilic cytoplasm. A lesion such as this one can also be mistaken for a Spitz nevus. Adequate immunohistochemistry can help resolve the differential diagnosis. It is important to remember that melanocytic lesions can also express CD68.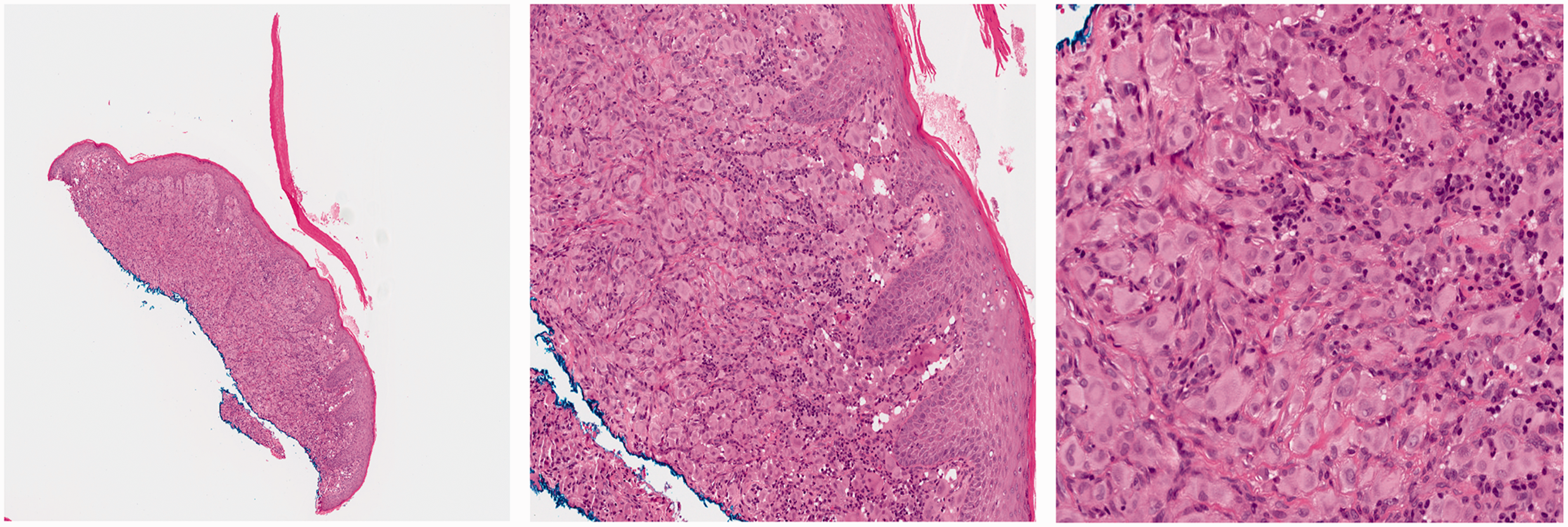
Systemic JXG should also be distinguished from ALK+ histiocytosis of childhood. ALK+ histiocytosis manifest with anemia, massive hepatosplenomegaly, and thrombocytopenia. The liver shows sinusoidal infiltration by histiocytic cells with folded nuclei, fine chromatin, small nucleoli, and voluminous eosinophilic cytoplasm with occasional hemophagocytosis. 385 Therefore, when diagnosing ECD and ALK, immunostain should always be performed.
JXG Family of Disorders
Benign cephalic histiocytosis
This rare disorder is localized to the skin, resolves spontaneously, and is considered to be an “early” form of JXG which affects young children, typically before 1 year of age, and has no gender predilection. Multiple tan-pink papules on the cheek, neck, and upper trunk are the clinical manifestations with sparring of the palms and soles, mucous membranes, and viscera. The papules measure up to 5 mm in diameter but may coalesce in a reticulate pattern. The clinical course is usually benign and self-limited; however, rare cases have been associated with diabetes insipidus and diabetes mellitus. The histopathologic features are those of JXG with diffuse dermal infiltrates of mononuclear histiocytes with ill-defined pale cytoplasm (Figure 20). Early lesions contain no cytoplasmic lipid, but later lesions become lipidized. Scattered lymphocytes and eosinophils may be seen. The immunophenotypic profile is identical to JXG.406–411
Benign cephalic histiocytosis. This is an uncommon histiocytosis in the family of xanthogranulomas. The lesion is ulcerated and shows rare multinucleated giant cells. Note the absence of lipidized histiocytes or definitive Touton-type giant cells.
Generalized eruptive histiocytosis
This rare disorder has been reported in both children and adults whose clinical presentation is the presence of recurrent crops of small, reddish papules symmetrically distributed on the trunk and extensor surfaces of the extremities without visceral involvement. Lesions develop rapidly and tend to resolve spontaneously with no residual findings or at most slight hyperpigmentation. It is known that lesions may occur several weeks after a bacterial or viral illness, but in adults, generalized eruptive histiocytosis may be associated with an underlying leukemia or lymphoma. Numerous mononuclear histiocytes, including spindled forms, are present within the upper to mid dermis and a perivascular distribution may be present. Scattered lymphocytes may be seen, but foamy cells or giant cells are generally absent.407,412–415 The clinical course is unpredictable since some cases resolve entirely and others progress to PNH or XD.
Progressive nodular histiocytosis
PNH mainly affects adults 40 to 60 years of age and is characterized by the presence of 2 types of skin lesions: multiple small, superficial yellow-brown papules and larger, deeper subcutaneous nodules with a predilection for the trunk. The papules measure up to 5 mm in diameter and are asymmetrically distributed over the body, although often sparing flexural areas. These lesions may become larger over time and can ulcerate and become painful; they often do not regress and may be progressive and deforming. Mucosal lesions may be present. Rarely, cases have been associated with acute myelomonocytic leukemia, chronic myeloid leukemia, or a hypothalamic tumor. A dermal infiltrate of histiocytes, predominantly spindle-shaped forms, and foamy cells (including Touton cells) are the histologic features. The infiltrate is present within a fibrous matrix, especially in older lesions, and, in some areas, a storiform pattern may be observed. It has been proposed that spindle cell xanthogranuloma is the solitary counterpart of PNH; presenting as yellow-brown papules or nodules on the head, trunk, and extremities of younger patients (20 to 40 years of age).416–419
Xanthoma disseminatum
XD is a rare disease in the JXG family that typically occurs in young adults with a male to female ratio of 2:1. Numerous reddish-brown papules, nodules, and plaques are symmetrically distributed on the trunk and upper extremities, including flexural sites; these lesions may become yellow and coalesce with time. Mucocutaneous sites, such as the oropharynx, larynx, conjunctiva, and cornea, are involved in up to 50% of cases. This disease has been associated with diabetes insipidus in almost 50% of cases; pituitary stalk involvement can also lead to hyperprolactinemia and hypopituitarism. The histologic findings are those of JXG (Figure 21).340,413,420
Xanthoma disseminatum. Another lesion in the family of JXG. There is a diffuse superficial dermal histiocytic infiltrate with a predominance of lipidized cells. Occasional Touton-type giant cells are noted.
Rosai–Dorfman Disease
RDD is a benign proliferative disorder of histiocytes. It is also termed sinus histiocytosis with massive lymphadenopathy. It is now included in the “R” group of histiocytosis. 340 Extranodal sites of involvement include the skin, oral and nasal cavities, respiratory tract, eyelid, and periorbital soft tissue. Systemic Rosai–Dorfman disease (SRDD) most commonly presents with fever and massive, painless, bilateral, cervical lymphadenopathy. Rosai and Dorfman first described the cutaneous manifestations of RDD among 10 patients, 7 of which were children. 421 Rarely, RDD can present solely with cutaneous lesions without systemic findings; this is known as cutaneous Rosai–Dorfman disease (CRDD).422,423 SRDD with cutaneous manifestations occurs in one-third of patients 424 while CRDD (disease without any other manifestations) is extremely rare. 425 Systemic disease favors young adult men and male children. 422 In contrast, CRDD favors adult white women, thus making it appear to be a distinct entity.422,423,425 As opposed to SRDD, CRDD is included in the group “C” of histiocytosis in the non-JXG family.
SRDD is benign and self-limited but can run a long relapsing and remitting course: it presents with fever and massive, nontender, bilateral cervical lymph node enlargement. Over 40% of patients will have extranodal involvement, the most common locations being the skin, soft tissues (including around the orbit), upper respiratory tract, salivary glands, CNS, and bone. Intrathoracic manifestations of SRDD are relatively common and present as mediastinal lymphadenopathy, airway disease, pleural effusion, cystic lung disease, and interstitial lung disease. These usually have a good prognosis. 426 Involvement of the lower respiratory tract, liver, and kidney portends a poor prognosis. 422
Immunological features are associated with 15% of patients with SRDD and include autoimmune hemolytic anemia and neutrophilia. 427 Such findings are a sign of poor prognosis.422,428–430 Cutaneous lesions associated with SRDD usually present on the eyelids and malar region. These are often nonspecific and appear as multiple, focal, erythematous, brown, or xanthomatous macules, papules, pustules, nodules, or plaques. Some of these lesions can be as large as 4 cm in diameter. In the rare cases of purely CRDD, they present as a central plaque or nodule with satellite papules or as multiple papules and nodules that coalesce into plaques.428,429
Histopathologically, there is an infiltrate of histiocytes with neutrophils, plasma cells, and scattered lymphocytes densely packed in the dermis (Figure 22). The histiocytes are polygonal and characteristically have an abundant, foamy, lightly eosinophilic cytoplasm with feathery borders and large vesicular nuclei and small nucleoli.
422
Emperipolesis (inflammatory cells and cell fragments engulfed within histiocytes) is a very important feature that characterizes RDD. The histiocytes are characteristically positive for S100 (Figure 22) and negative for CD1a.
431
Multinucleated histiocytes may present as aggregates in Touton-like giant cell formation. In the cutaneous lesions, plasma cells may contain Russell bodies and present with associated xanthoma cells, fibrosis, and necrosis. CRDD differs histologically from nodal disease in that there is a greater degree of fibrosis, fewer histiocytes, and less emperipolesis. As a result, CRDD can be overlooked if the index of suspicion for the disease is low, perhaps contributing to its rare diagnosis.
432
The histology may also demonstrate a mixed septal and lobular panniculitis.
423
Many cases of CRDD will often elicit a “lymph node in skin” appearance. Histopathologic samples that are suspicious for RDD or another form of histiocytosis are stained with S100, CD163, CD68, and CD1a. Positive staining of histiocytes with S100 and CD68 with a negative CD1a stain suggests RDD (Figure 22). Positivity of all 3 stains suggests LCH and/or indeterminate cell histiocytosis. RDD can also have increased IgG4+ plasma cells.433–435 However, the significance of this finding is not yet known. Both IgG and IgG4 stains should always be performed together, in order to properly evaluate the ratio of IgG4 to IgG+ plasma cells.
Cutaneous Rosai–Dorfman disease (CRDD). A dense infiltrate of histiocytes, lymphocytes, and plasma cells is seen. The histiocytes have an epithelioid appearance. Many of them show engulfment of other inflammatory cells, “emperipolesis.” High-power magnification is shown for emperipolesis. The histiocytic cells of RDD are positive for CD163 and S100.
In 2 families reported to have hereditary RDD, biallelic germline mutations in SLC29A3 have been found with phenotypic expression in the CNS, eye, inner ear, and epithelial tissues. SLC29A3 encodes an intracellular equilibrate nucleoside transporter (hENT3). 436 RDD is mostly sporadic, but inherited cases can occur. Mutations and large deletions of greater than 50 base pairs in mitochondrial DNA have rarely been identified in tissue samples of CRDD. 437 A closely related entity, the “H syndrome,” has been associated with mutations on the SLC29A3 gene. The syndrome is characterized by numerous cutaneous findings, including hyperpigmentation, hypertrichosis, hepatosplenomegaly, hearing abnormalities, hypogonadism, low height, hyperglycemia, and hallux valgus. The cutaneous findings in this process mimic those of RDD with a CD68+, S100+, CD1a−, histiocytosis with emperipolesis, and abundant plasma cells.438–440
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.