
Abstract
Langerhans cell histiocytosis (LCH) and juvenile xanthogranuloma (JXG) both belong to the histiocytosis group of disorders, which have varied prognostic and clinical significance. Their normal cellular counterparts share a common CD34-positive hematopoietic stem cell precursor that matures along CD14-negative or -positive pathways. Rare cases of LCH and JXG show overlapping findings, suggesting that this divergent maturation is not irreversible. We report a case of an infant diagnosed with cutaneous LCH shortly after birth. Two years after diagnosis, a recurrent lesion in his external auditory canal contained lipidized cells with CD68 and factor XIIIa expression and lacked Birbeck granules and CD1a, consistent with JXG. Our case and previous reports of this phenomenon raise the question of a histogenic relationship between LCH and JXG, and we speculate that the lesional cells of origin are subjected to therapy-induced modulation that results in a varied differentiation.
INTRODUCTION
Histiocytic disorders are categorized based on their normal counterpart and proposed cell of origin. Langerhans cell histiocytosis (LCH) and juvenile xanthogranuloma (JXG) were both believed to be myeloid-derived, dendritic cell (DC)-related disorders [1,2]. However, more recently studies have shown that the lesional cell of origin in JXG may, in fact, be the dermal macrophage and not DC, as previously thought [3]. According to the current paradigm, CD34-positive hematopoietic stem cells mature into cells with multilymphoid progenitor potential, which further differentiates along 2 major pathways of divergent maturation attributed to their cytokine environment: CD14-/CD1a+ DCs and CD14+/CD1a- macrophages [4,5]. CD14- cells expressing CD1a, S100, and langerin (CD207) represent the lesional cells of LCH, the LCs. CD14+ cells that lack CD1a and langerin but express factor XIIIa, CD68, and CD163 are postulated to be the precursors of JXG, the dermal macrophages. However, this hypothesis has been highly debated, and other normal counterparts, such as plasmacytoid monocytes, have been proposed as the precursors of JXG [6,7]. Further complexity is added by rare, perplexing reports of LCH and JXG presenting in the same patient, either as coexisting or metachronous lesions [8–14]. We report 1 such case in which JXG was diagnosed almost 2 years after the initial development of LCH in a young child.
CASE REPORT
A 2-year-old boy presented with fever and a granulation tissue-like mass in his right external auditory canal. Physical examination revealed a poorly defined tympanic membrane obscured by the lesion whose limits could not be reached by otoscopy. Computerized tomography revealed erosion of the incus and opacification of the right middle ear, mastoid, and paranasal sinuses. The inner ear was uninvolved. A complete blood count showed a leukocyte count of 7 × 109/L, a normal differential count, a hemoglobin concentration of 12.2 g/dL, and a platelet count of 448 × 109/L. Ear examination under general anesthesia was accompanied with a biopsy of the auditory canal mass.
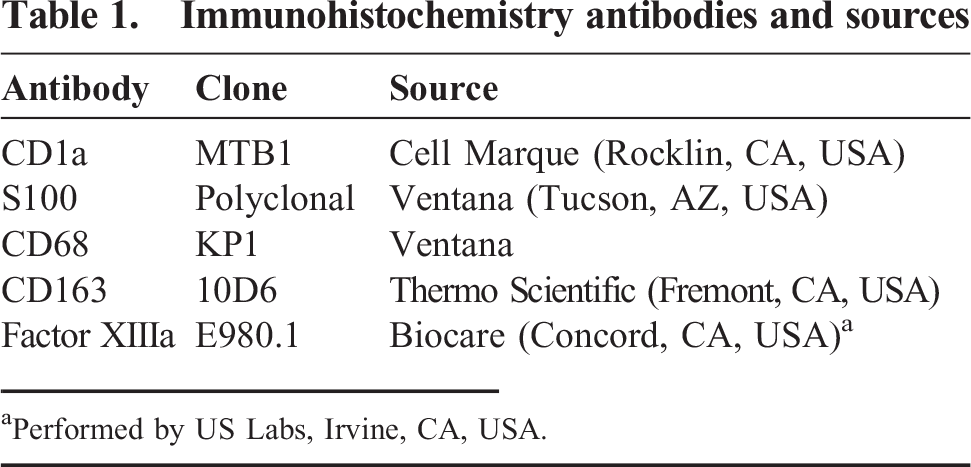
The patient's prior history was significant for a rash involving his torso, upper extremities, scalp, and soles, noted by his mother soon after birth. He was born by caesarian section and has a normal nonidentical twin. He was admitted at 8 weeks of age, with a complete blood count showing a leukocyte count of 11.1 × 109/L, a hemoglobin of 12.2 g/dL, a slightly elevated platelet count of 375 × 109/L, an absolute neutrophil count of 0.3 × 109/L, and a relative lymphocytosis. A shave biopsy of one of the cutaneous lesions contained numerous scattered atypical grooved large cells and abundant eosinophils within the epidermis and superficial dermis (Fig. 1). The atypical cells were positive for CD1a and S100 and showed variable staining for CD163 (Table 1). The morphologic findings and immunohistochemical stains were consistent with a diagnosis of LCH. However, langerin immunohistochemical staining and electron microscopy were not performed. Because CD1a and S100 staining has previously been reported in cases of JXG [13], we performed electron microscopy in retrospect on a section of prior paraffin-embedded biopsy material and confirmed the presence of Birbeck granules (Fig. 1). A skeletal survey was normal. Chemotherapy consisted of oral corticosteroids only, with a good early response and clearing of the lesions. However, the disease relapsed when the patient was 9 months of age, and chemotherapy with vinblastine and prednisone were started. Once again, the lesions showed a good response, followed by a 2nd relapse at 17 months, and a skeletal radiologic survey and computed tomographic scan disclosing multiple lytic lesions involving the calvarium, left maxilla, and mandible. A 2nd round of chemotherapy with vinblastine, prednisone, and methotrexate was started, and the bony lesions resolved over several months, leading to the current apparent relapse at 2 years of age. During this time, there was never any documented radiologic evidence or symptoms of tumor involving the external ears.
Immunohistochemistry antibodies and sources
Performed by US Labs, Irvine, CA, USA.
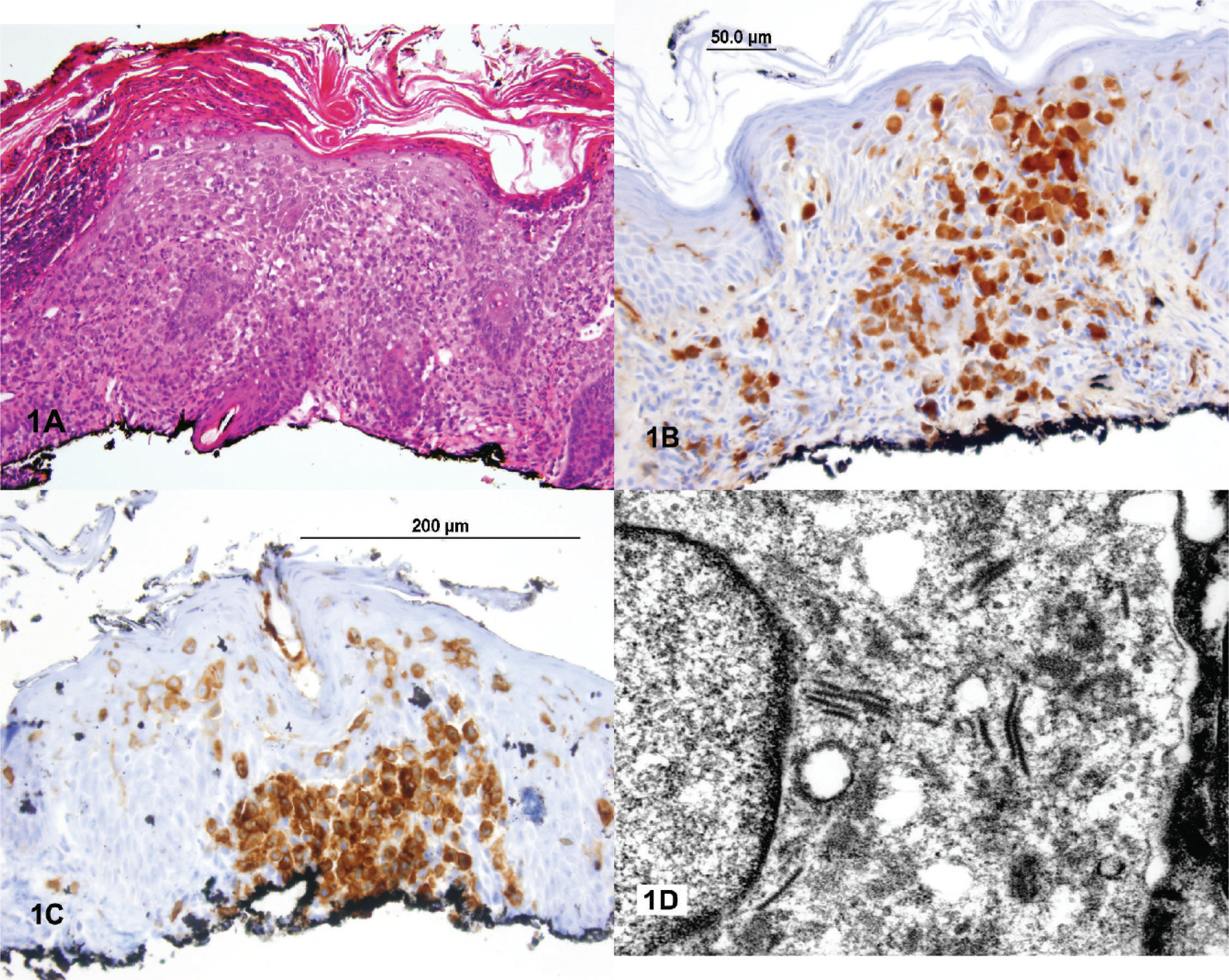
The current biopsy consisted of portions of squamous mucosa containing an intense mixed infiltrate of inflammatory cells, including neutrophils, occasional eosinophils, and prominent histiocytes. Some of the cells had lipidized cytoplasm. Nuclear grooves were not apparent. A few scattered multinucleated giant cells were also present (Fig. 2). Special stains were negative for acid-fast bacilli, bacteria, and fungus. Immunohistochemical stains revealed cells strongly positive for CD68 and factor XIIIa. Occasional CD1a- and S100-positive cells were identified, but the primary lesional cells were clearly negative for both markers. No Birbeck granules were identified by electron microscopy, which showed nonspecific findings. These findings were considered most consistent with a diagnosis of JXG rather than LCH.
DISCUSSION
Our case supports the previously described complex relationship between LCH and JXG, which are both considered disorders arising from CD34+ hematopoietic stem cells but with a divergent lineage of maturation. Langerhans cell histiocytosis arises as a clonal proliferation of LCs and specialized DCs present in the skin and mucosal sites. However, whether it arises from transformed resident LC or bone marrow–derived DC precursor is uncertain, with cell-specific gene expression study in LCH favoring the latter hypothesis [15]. Langerhans cells are identified by the expression of CD1a, S100, and langerin (CD207) and the presence of Birbeck granules [2,15]. Langerhans cell histiocytosis ranges from a single lesion with spontaneous remission to a rapidly progressive multisystem disorder with fatal outcome; yet its reactive or neoplastic nature is largely unsettled [16].
Macrophages are the presumed normal counterpart of JXG and express CD68/CD163 and factor XlIIa. However, Kraus and colleagues [6] raised the possibility that circulating plasmacytoid monocytes may represent the immediate precursors of the DCs that give rise to JXG. This hypothesis was based on number of observations, including the dearth of factor XIIIa–positive cells in normal control tissues and the occurrence of JXG in locations other than skin. They further suggested that the cells of origin of JXG are normally circulating monocytes that migrate to injured/inflamed tissues under the correct cytokine stimulus. However, significant evidence is lacking to further support this hypothesis. Hoeger and colleagues [11] also speculated a reactive nature of the development of JXG. Conversely, recently reported evidence for the clonal nature of JXG and reports of successful treatment of JXG with LCH-directed chemotherapy also argue against its inflammatory/reactive nature [7,17–19]. Recently, Zaba and colleagues [3] demonstrated that normal human dermis has CD14+/CD68+/CD163+ macrophages that also express factor XIIIa, and the JXG lesional cell phenotype is also similar to a dermal macrophage rather than DC [4].
Of note, there was no documented involvement of the patient's right auditory canal during his prior treatment and follow-up appointments for the primary diagnosis of LCH. The possibility that a xanthomatous lesion appearing after LCH may represent a late stage of a previously unnoticed and later involuted or treated LCH exists. However, it is highly unlikely considering that the patient had multiple diagnostic and follow-up computed tomographic scans of his head and neck region and a regular follow-up physical examination with no identified lesions involving his right ear in course of his primary illness.
The linked origin of macrophages and DCs from a common precursor macrophage-DC progenitor cell is demonstrated with capabilities to differentiate into CD14+/CD1a- macrophages or CD14-/CD1a+ LCs under the correct cytokine stimulus [4,20,21]. Thus, an alternative possibility is the presence of trans-differentiated precursor transitional cells that maintain the capability of differentiating towards either lineage. The etiology of LCH and JXG is uncertain; however, their recurrent clinical association indicates more than a chance occurrence. The concept of lineage-specific maturation driven by the cytokine environment is well accepted, and the lesions arising from disordered maturation generally retain distinct phenotypes. However, such distinction may not always hold true, as evidenced by our case and other cases of coexisting LCH and JXG. One may further speculate that modulation of cytokines as a result of LCH chemotherapy agents may result in the development of JXG subsequently later in time and that the divergent maturation of the common precursor of LCs and macrophages is not completely irreversible. Moreover, coexisting LCH and JXG may occur without any intervention or modulation of the cytokine environment by chemotherapeutic agents, such that a divergent but concurrent clonal differentiation of macrophage-DC progenitor cell also remains a matter of conjecture.
Notably, the expression of CD1a is not specific for LCH but has also been variably reported in some lymphoid and myelomonocytic malignancies, which can infiltrate skin and other organs [22,23]. Isolated case reports documenting positivity for CD1a in JXG, sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease), and indeterminate cell histiocytosis are also evident; however, it has never been confirmed by any larger series of study [13,24–27]. Expression of langerin and Birbeck granules has also been recently shown in cells other than LCs, hence disputing the unique and specific nature of these findings once attributed only to epidermal LCs [2,16].
In summary, we present a case of 2 apparent manifestations of histiocytosis in the same patient, whose findings suggest a need to re-evaluate the histogenesis of LCH and JXG. A common macrophage-DC progenitor cell may represent the missing link between the 2 disorders.
Footnotes
ACKNOWLEDGMENT
We thank members of the University of Oklahoma Medical Center Histopathology Laboratory for their assistance in performing the relevant immunohistochemistry staining and providing the necessary information.