
Abstract
The numerical solution of the Kadanoff-Baym nonlinear integro-differential equations, which yields the non-equilibrium Green’s functions (NEGFs) of quantum many-body systems, poses significant computational challenges due to its high computational complexity. In this work, we present efficient implementations of a numerical method for solving these equations on distributed-memory architectures, including many-core CPUs and multi-GPU systems. For CPU-based platforms, we adopt a hybrid MPI + OpenMP programming model to exploit both inter-node and intra-node parallelism. On GPU-accelerated systems, we implement the method using two distinct approaches: MPI + OpenACC and MPI + CUDA FORTRAN. Several optimization strategies are employed to enhance GPU performance, including techniques to maximize computational resource utilization and minimize the overhead associated with kernel launches and memory management. Although OpenACC is easy to use, CUDA FORTRAN provides more advanced features for configuring and managing multiple levels of concurrency, while also simplifying memory allocation and data movement between host and device. This flexibility translates into significant performance improvements. For a representative problem with 1024 k-points, the GPU implementations accelerate the dominant self-energy calculation by more than two orders of magnitude relative to the CPU implementation, with speedups of 103 − 141× using OpenACC and 123 − 175× using CUDA FORTRAN. The collision-integral calculation is also substantially accelerated, achieving speedups of 36 − 45× with OpenACC and 45 − 52× with CUDA FORTRAN. We compare the performance of the three implementations and show that CUDA FORTRAN consistently outperforms OpenACC, achieving up to a 1.34× additional speedup over the best OpenACC configuration for the self-energy calculation. Furthermore, both CPU and GPU versions exhibit excellent strong and weak scaling, confirming the scalability and efficiency of our approach for large-scale NEGF computations.
1. Introduction
Simulating the dynamics of a nonequilibrium quantum many-body system with a large number of particles N is computationally challenging due to the exponential scaling of the number of degrees of freedom in the many-body wavefunction with respect to N. However, many properties of a quantum many-body system can be ascertained by working with its observables such as the single-particle Green’s function G(r, t, r′, t′), which can be interepreted as the probability of detecting a particle at the position r and time t when a particle is created at position r′ and time t′. For simplicity, we will leave out the spatial coordinates in G below. For systems that are out of equilibrium, the non-equilibrium Green’s function (NEGF) is a two-time function G(t, t′) that satisfies an equation of motion known as the Kadanoff-Baym equation (KBE) (Kadanoff and Baym, 1962; Stefanucci and Van Leeuwen, 2013; Kadanoff and Baym (2018); Fetter and Walecka, 1971), which is a set of nonlinear integro-differential equations defined on a contour
The KBEs have the form (Lipavskỳ et al., 1986; Kadanoff and Baym, 2018).
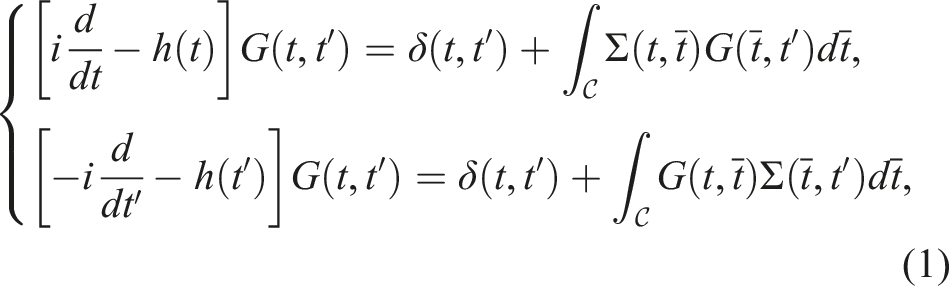
The numerical solution of equation (1) requires propagating G(t, t′) on a two-time grid. The time evolution procedure is computationally expensive due to the coupled nature of the equations, the nonlinearity of the integro-differential equation, and the need to evaluate the integral term, often referred to as the collision integral, at each time step in addition to the self-energy evaluations. These self-energy terms are dependent on the Green’s function, and their evaluation requires several tensor contractions.
Fortunately, multiple levels of concurrency exist in both the self-energy and the collision integral calculations. The algorithmic concurrency allows us to implement these calculations efficiently on many-core CPUs using the MPI + OpenMP programming model. However, performing a long-time NEGF dynamics simulation on a many-core CPU based distributed memory high performance computer can still take a prohibitively long wall clock time.
In recent years, high-performance computers equipped with graphics processing unit (GPU) accelerators have become widely available. GPUs possess significantly more hardware threads than CPUs and offer much higher memory bandwidth. They appear to be the natural computing platform for carrying out NGEF simulations.
However, there are some unique features of GPUs that make it challenging to take full advantage of the computational resources available on such an architecture. To efficiently utilize multiple GPUs for solving KBEs, the original MPI + OpenMP implementation framework must be extended to support an offloading programming model. Achieving optimal performance in this context requires careful optimization to ensure maximum resource utilization and minimal computational overhead. Open standards such as OpenACC and OpenMP target directives offer portability across hardware platforms, while vendor-specific models like CUDA FORTRAN can provide higher performance on supported devices. Choosing between these approaches involves balancing portability and performance requirements.
We discuss the changes we need to make in an MPI + OpenACC implementation of the self-energy and collision integral calculations in order to achieve satisfactory performance on NVIDIA GPUs. We show how performance profiling tools such as NVIDIA’s Nsight Compute can be used to guide performance optimization. Although OpenACC is relatively easy to use, it has some limitation on resource allocation and utilization. We show that CUDA FORTRAN can provide more flexibility and allow us to fine tune the performance by managing how thread blocks and threads are configured and utilized to achieve multiple levels of concurrency as well as reduce memory allocation and host-device data transfer overhead.
The paper is organized as follows. In Section 2, we describe the main computational tasks in the numerical solution of the KBE and examine the different levels of concurrency available in the main computational kernels. In Section 3, we discuss how these computational tasks can be parallelized on distributed-memory many-core CPUs using a hybrid MPI + OpenMP implementation, and we demonstrate the parallel scaling of this CPU-based implementation. In Section 4, we describe how OpenACC can be used to replace OpenMP and enable the KBE solver to run efficiently on NVIDIA GPUs. In Section 5, we present a CUDA FORTRAN implementation and show how explicit control over kernel launches, thread-block organization, reductions, and device memory management can further improve GPU performance. In Section 6, we present computational experiments on the Perlmutter supercomputer, compare the performance of the MPI + OpenMP, MPI + OpenACC, and MPI + CUDA FORTRAN implementations, and examine both strong and weak scaling behavior. We also study the sensitivity of GPU performance to OpenACC gang/vector sizes and CUDA FORTRAN grid/block configurations. Finally, Section 7 summarizes the main findings and discusses the implications of GPU acceleration for large-scale NEGF simulations.
2. Computational kernels
In this section, we use a specific model problem to illustrate the main computational kernels used in a numerical time evolution scheme for solving the KBEs given in equation (1). The model problem is a so-called two-band Hubbard model (Hirsch, 1985; White et al., 1989; Macridin et al., 2005; Werner et al., 2009; Pudleiner et al., 2016), which is a simplified model that describes the essential physics of interacting electrons on a N-site lattice. The Hamiltonian for such a model is defined as
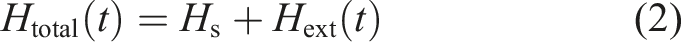
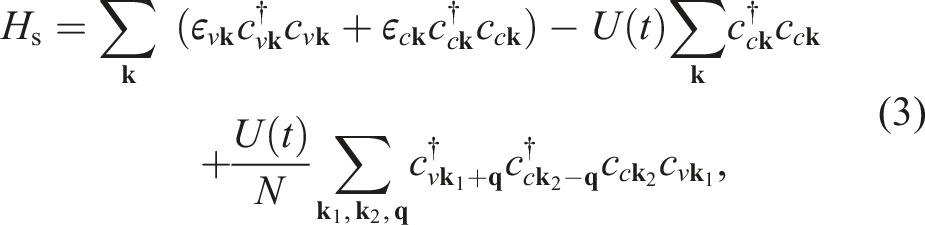
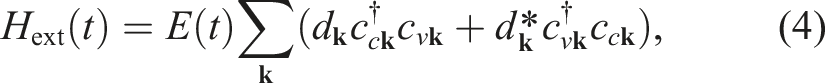
The KBEs associated with such a model problem can be written as
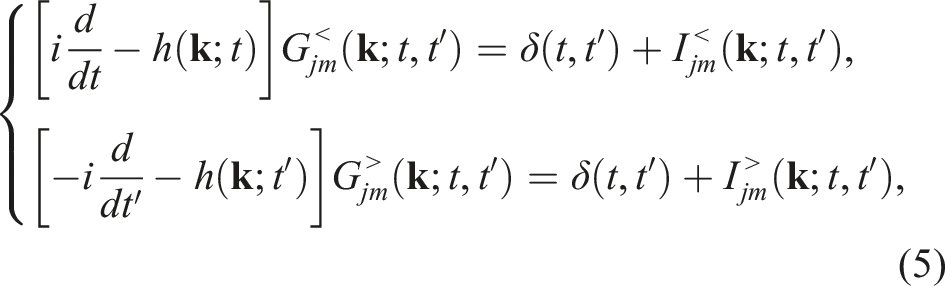
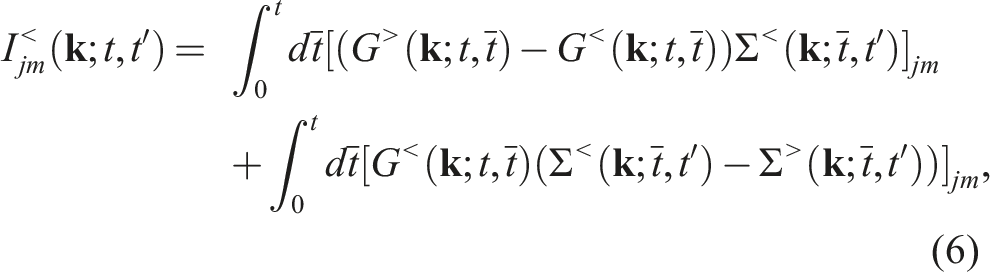
Although the integro-differential equation (5) may appear to be completely independent for different
When the second-Born approximation is used to define the self-energy terms, we can write
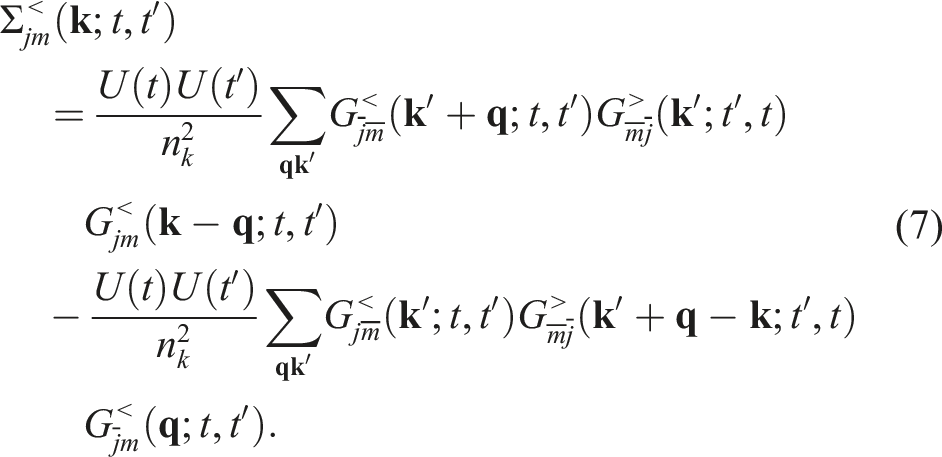
Similar expressions can be written for
The numerical method we use to solve equation (5) advances G≶(t, t′) along both the t and the t′ directions. If we let

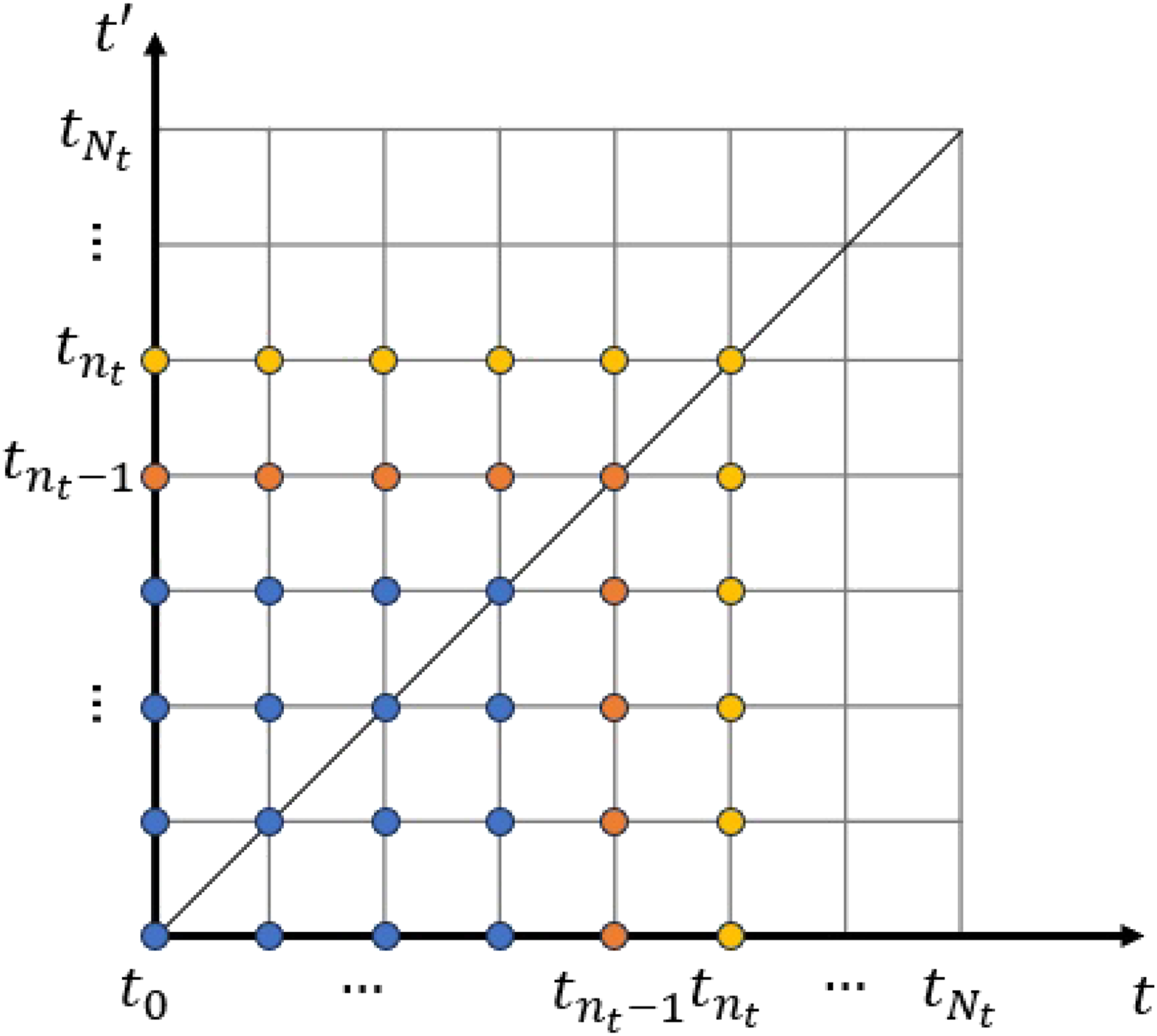
At the n
t
-th time step, when advancing from • Evaluate the single-particle Hamiltonian • Evaluate the self-energies • Evaluate the collision integral • Numerically evolve Illustration of the evaluation at the n
t
-th time step for Σ≶ and I≶. The blue dots represent values computed at previous time steps, and the orange dots give the 2n
t
− 1 pairs of (t, t′) at which the values of Σ≶ and I≶ are computed at the n
t
-th time step.
A similar set of operations are performed to advance
Note that the self-energy Σ< defined in equation (7) is the difference of two terms. We denote the first and second terms by
We compute
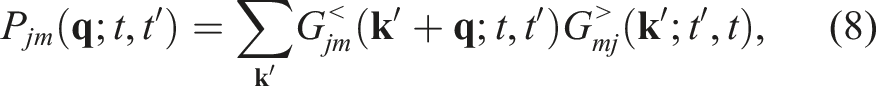
This step contracts
In the second step, we contract
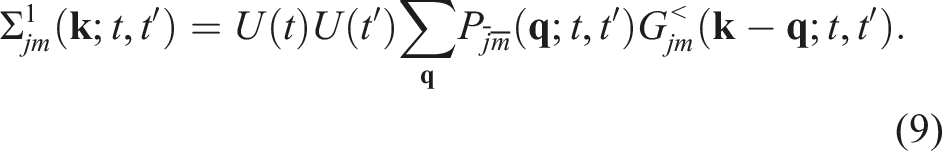
This contraction also costs
By breaking the contractions over
However, such a reduction in complexity is not applicable to

The collision integral given in equation (6) can be evaluated as the sum of two terms
Since the evaluations of I1 and I2 are similar, once we have G≶ and Σ≶ computed and stored, we will only discuss how I2 is computed below.
The main computation performed in the evaluation of I2 is the convolution of
Algorithm 1 shows that we need to perform at least n
t
self-energy and collision integral calculation in order to obtain approximations to
3. MPI + OpenMP implementation
The separation of the KBEs (5) by k-points for the two-band Hubbard model as shown in equation (7) naturally suggests that we can distribute the time evolution of The main steps in the outermost loop of the KBE solver.
We use CrayPat to analyze the performance of the MPI implementation. In a run that propagates the Green’s function for 10 time steps on 1024 MPI ranks with n k = 1024 k-points, the profiler shows that the self-energy computation takes over 95% of the total time. This observation is consistent with our previous cost analysis that suggests the self energy evaluatation would dominate the entire computation when n t ≪ n k . Therefore, in the following, we focus first on investigating the performance of the self-energy computation.
In Figure 3, we show a code snippet for computing The MPI implementation of 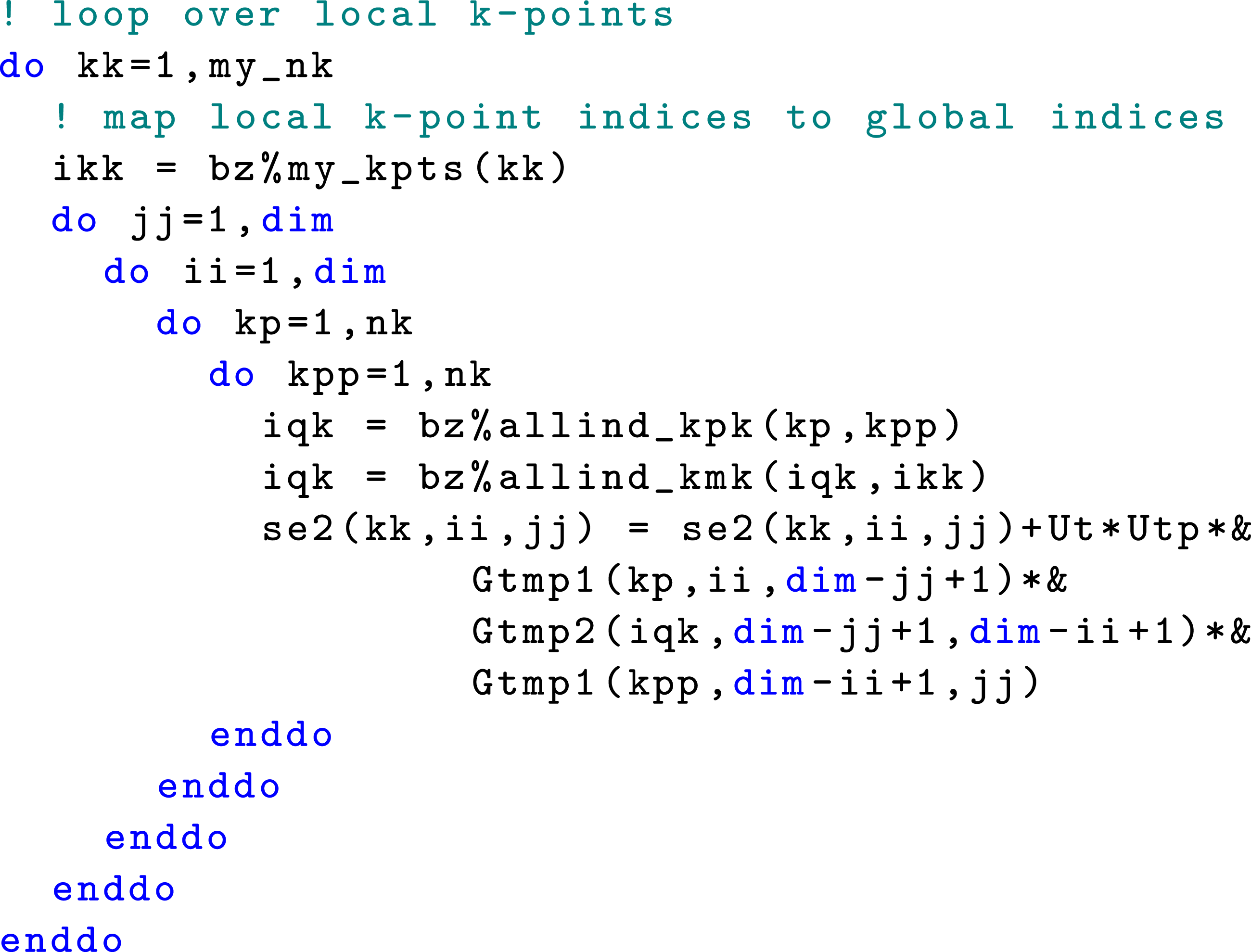
The nested loop structure shown in Algorithm 2 and Figure 3 indicates that within each MPI rank, additional levels of concurrency for the self-energy evaluation can be exploited by using OpenMP.
Because the number of bands (2) in the model Hamiltonian is far fewer than the number of threads within each MPI rank (on a manycore compute node) compared to the number of k-points, we reorder the nested loops in Figure 3 to move the The hybrid MPI + OpenMP implementation of the 
It is possible to implement the contractions of the Green’s function in (7) as basic linear algebra operations (BLAS) and call an optimized BLAS library. For example, for each t, t′, j and m, we can first reshape
Figure 5 shows the strong scaling performance for the computation of the non-equilibrium Green’s function. These calculations have been performed on the Perlmutter CPU partition at NERSC (National Energy Research Scientific Computing). Each node is composed of 2 AMD EPYC 7763 (Milan) with 64 cores per CPU. In this experiment, we set the number of k-points to The performance of the MPI and the hybrid MPI + OpenMP implementation to the program with varying number of MPI ranks. In the test problem, n
k
= 1024, and we compute the first 10 time steps.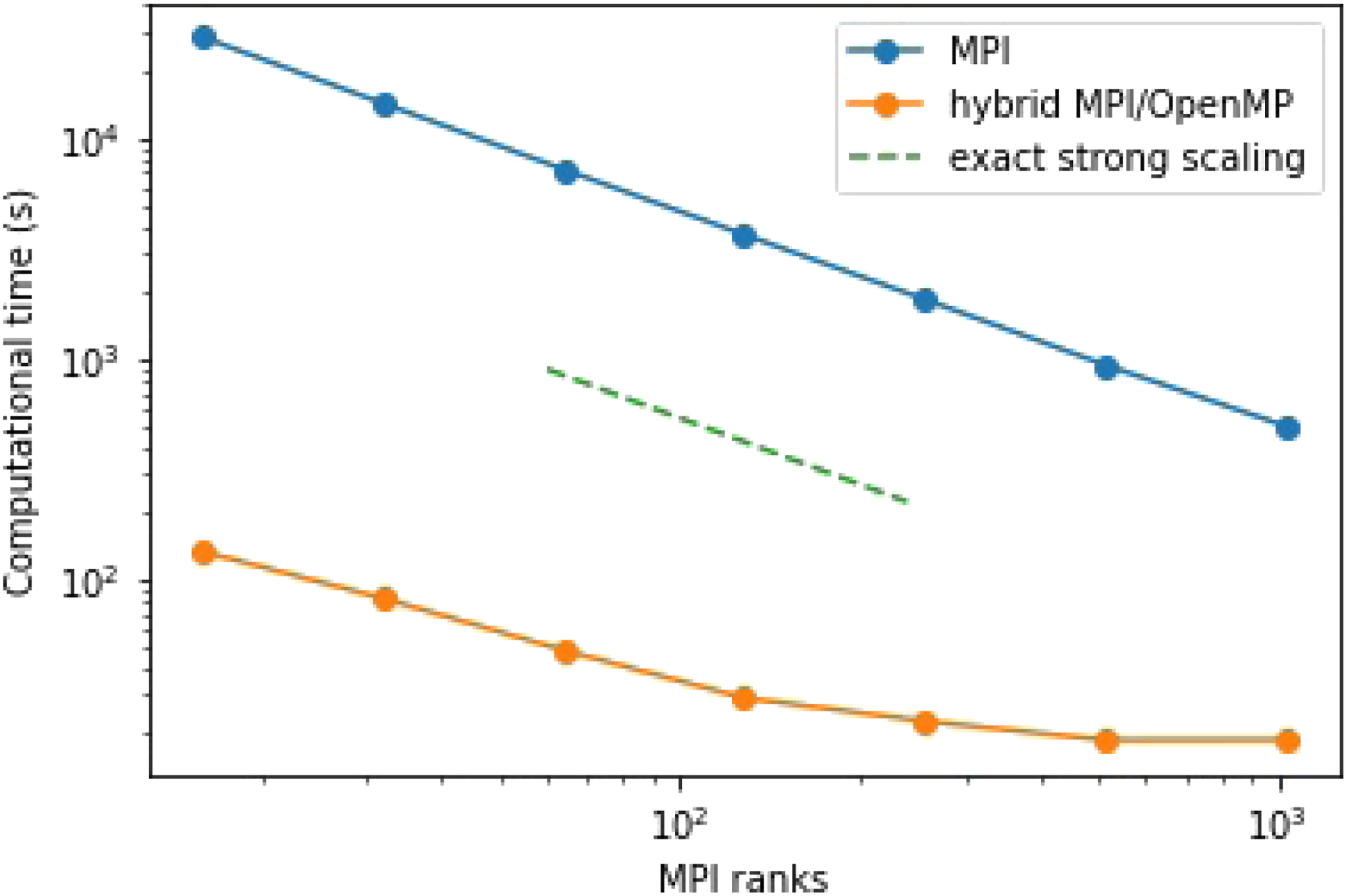
4. GPU implementation with OpenACC
Although the MPI implementation exhibits nearly perfect strong scaling, Figure 5 shows that the wall clock time required to evolve the Green’s function for 10 time steps already exceeds 8 minutes even with 1024 MPI ranks. As mentioned before, the calculation of the self-energy and the collision integral takes over 95% of the total computational time. The use of hybrid MPI + OpenMP parallelization allows us to reduce the computation time by a factor of 26 on 1024 MPI ranks with 64 threads per MPI rank. However, when the number of time steps increases, the self-energy and collision integral evaluation will become proportionally more time-consuming. To obtain a trajectory with N t = 500 time steps, the hybrid MPI + OpenMP implementation uses more than 3 hours to compute the self-energy and about 2 hours to evaluate the collision integral.
To further improve the throughput of NEGF computation, we investigated the possibility of running the KBE solver efficiently on high performance computers equipped with accelerators. In this work, we focus on enabling the KBE solver to run efficiently on NVIDIA graphics processing units (GPUs).
Compared to mainstream multi-core CPUs, GPUs have a larger number of primary execution units which are the Streaming Multiprocessors (SMs). Each SM contains resources to execute thousands of SIMT (Single Instruction, Multiple Thread) threads organized into thread blocks and warps. The SIMT execution model and the availability of a large amount of high bandwidth shared memory and local cache make GPUs ideal for computations that have high arithmetic intensity and regular memory access patterns.
To leverage the massive parallelism available on GPUs, we need to design the solver to map concurrent tasks performed in the KBE solver within an MPI rank to different threads or thread blocks excecuted simultaneously on a GPU device. On NVIDIA GPUs, the easiest programming model that allows us to achieve such a mapping is the OpenACC directive-based open standard programming model (OpenACC-Standard.org, 2022; Chandrasekaran and Juckeland, 2017), which is an open source parallel programming standard.
Although a GPU node has much higher peak performance than a CPU node, there exist a few challenges that can hinder the optimal execution of code on a GPU. In particular, repeated memory allocations on the GPU device and excessive data movement between the host CPU and the GPU device can introduce a significant amount of overhead. The execution of a computational task on the GPU requires a kernel launch. A large number of repeated kernel launches can also degrade the performance of the overall computation. Furthermore, a sub-optimal memory access pattern that results in a memory bank conflict can also contribute to sub-optimal performance of the code.
In this section, we describe how to overcome these challenges when OpenACC directives are used to offload the Σ2 and I2 calculations onto NVIDIA GPUs. We can use OpenACC directives to manage memory access and map computational work to multiple thread blocks and threads to achieve optimal utilization of GPU resources.
We will demonstrate various performance improvement resulting from the use of OpenACC directives through a test problem that uses n k = 1024, n t = 500. The problem is solved on a single node of the Perlmutter GPU partition, namely using 4 MPI ranks each comprising one NVIDIA A100 GPU.
4.1. Exploiting GPU concurrency by loop fusion and reordering
OpenACC provides a Using OpenACC 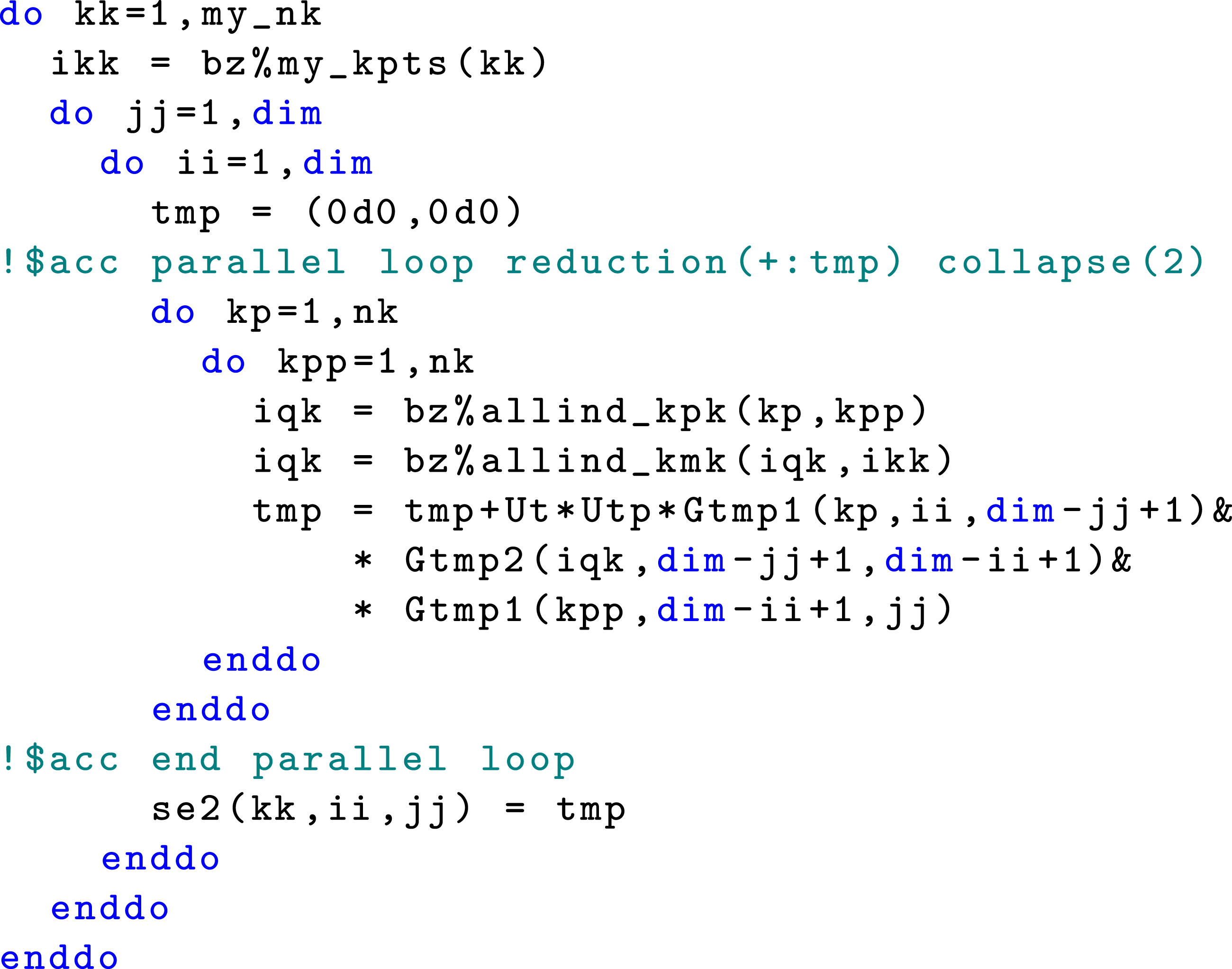
Another way to leverage the GPU architecture is to exploit multiple levels of concurrency that exists in the nested loops in Algorithm 2. This can be achieved by using the The OpenACC implementation for computing the 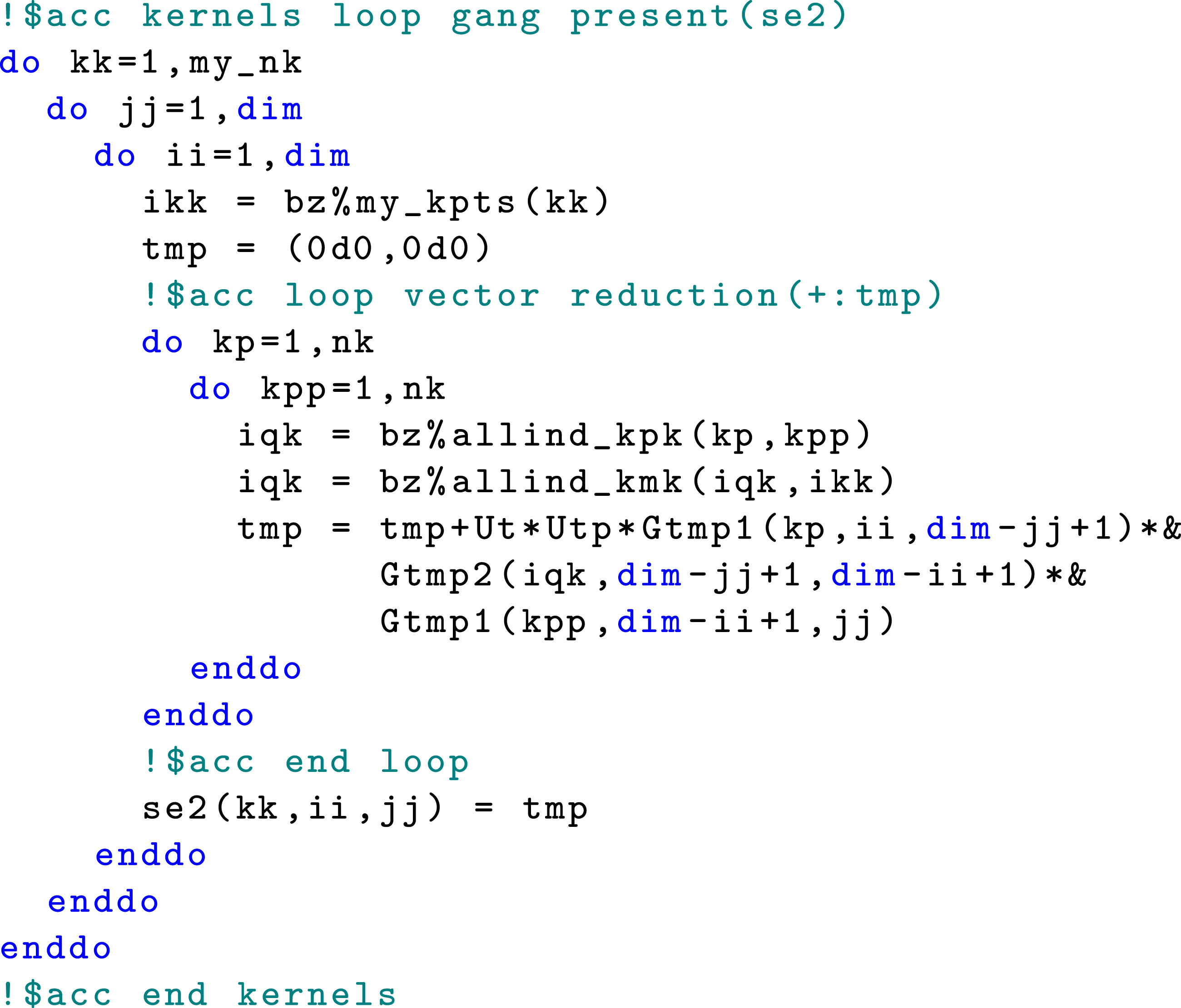
4.2. Updating self-energy using a single kernel launch
In a naive implementation of Algorithm 1 that loops over the j and ℓ indices to update The main loop for computing the self-energy.
Because each Kernel launches inside the subroutine 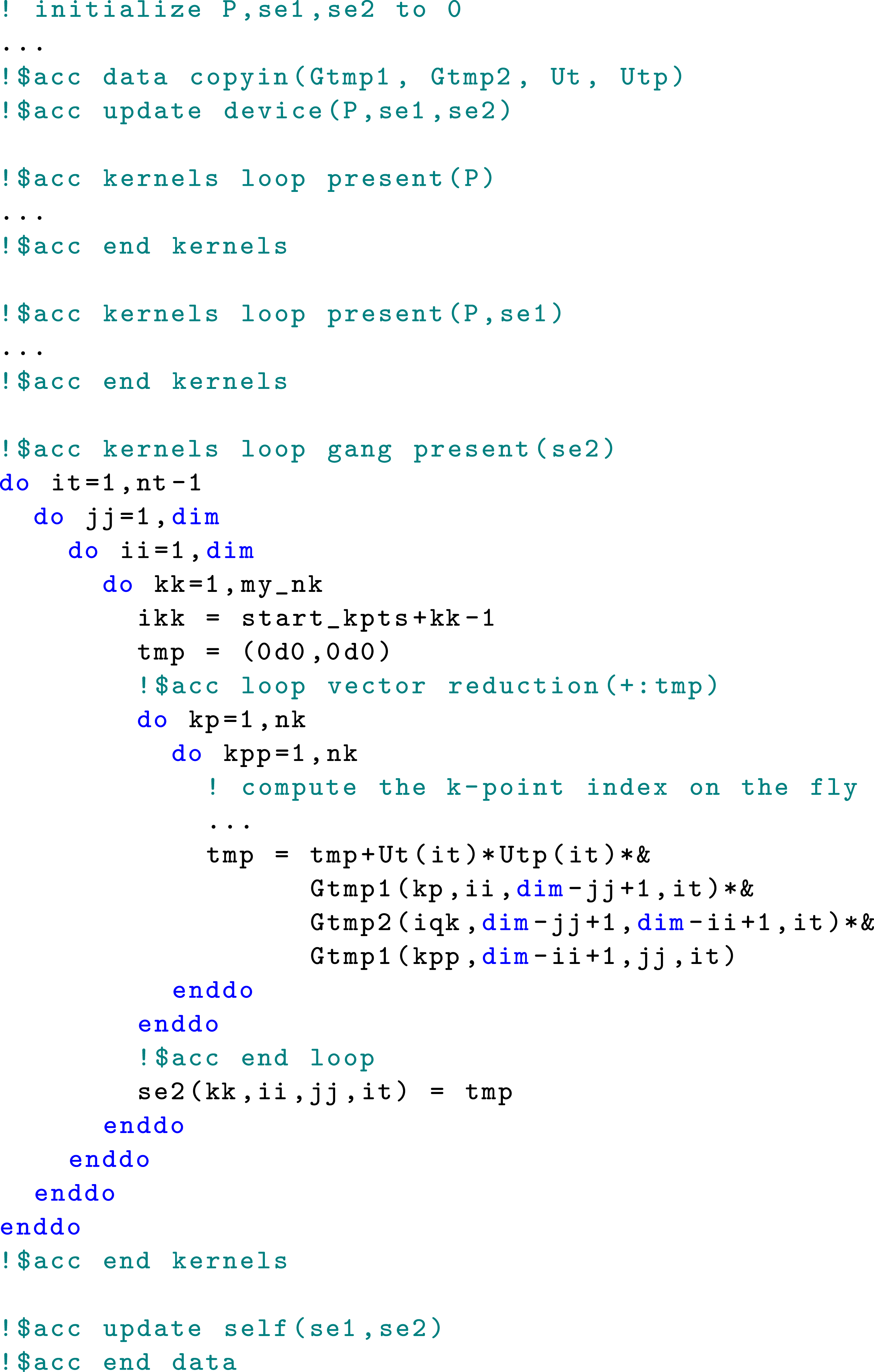
Figure 10 shows how this subroutine is called. Note that instead of passing a single time snapshot of the The calls to the self-energy computation subroutine with the code restructure.
A similar code restructuring is used to implement a version of collision integral computation that uses a single kernel launch. We omit the corresponding code snippets here for brevity. This approach allows us to achieve high concurrency and considerably reduces the amount of time spent in the self-energy and collision integral calculations. For example, using this restructured code results in a 90% performance improvement in the self-energy calculation compared with the version that implements only the loop reordering shown in Figure 7 for the model problem described in Section 2.
4.3. Reducing memory allocation and data movement overhead
There are several allocatable arrays used to hold intermediate results in the self-energy and the collision integral calculations. For example, in the MPI + OpenMPI version of the self-energy computation, the temporary array
To reduce this overhead, we modified the code to allocate these arrays only once on the host, prior to calling the self-energy calculation subroutine. Once allocated, these arrays are immediately copied to the GPU device. There is no need for repeated allocation and data transfer inside the loop over t or t′.
The self-energy computational kernels are launched inside the subroutine
Moreover, since we use
For the test problem, optimizing memory allocation and data transfer between the host and device yields an additional 8.8% performance improvement in the self-energy computation. Similar techniques can be used to reduce memory allocation and data transfer overhead in the evaluation of the collision integral.
4.4. Trading on-the-fly index calculation for reduced irregular memory access
In the OpenMP implementation shown in Figure 4, the k-point index
In the model problem, we sample the k-points uniformly on the interval
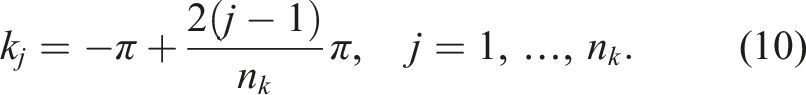
The sum of two sampled k-points falls within
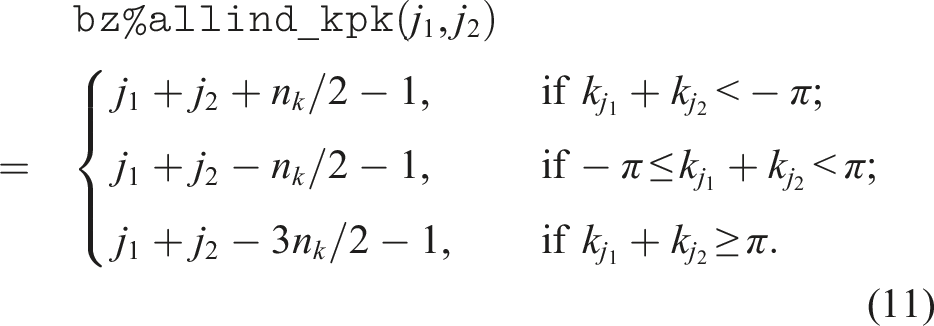
A similar expression can be used to obtain the index of
By evaluating these expressions on the fly, we can avoid unnecessary memory access that impedes concurrent executions of the self-energy and collision integral calculations. This optimization gives an additional 3.4% improvement in the performance of self-energy computation.
4.5. Performance assessment
The benefits of all strategies we discussed above are displayed and compared in Figure 11. We can see that loop reordering and the use of The wall clock time (seconds) used by different variants of the OpenACC implementation for the model problem.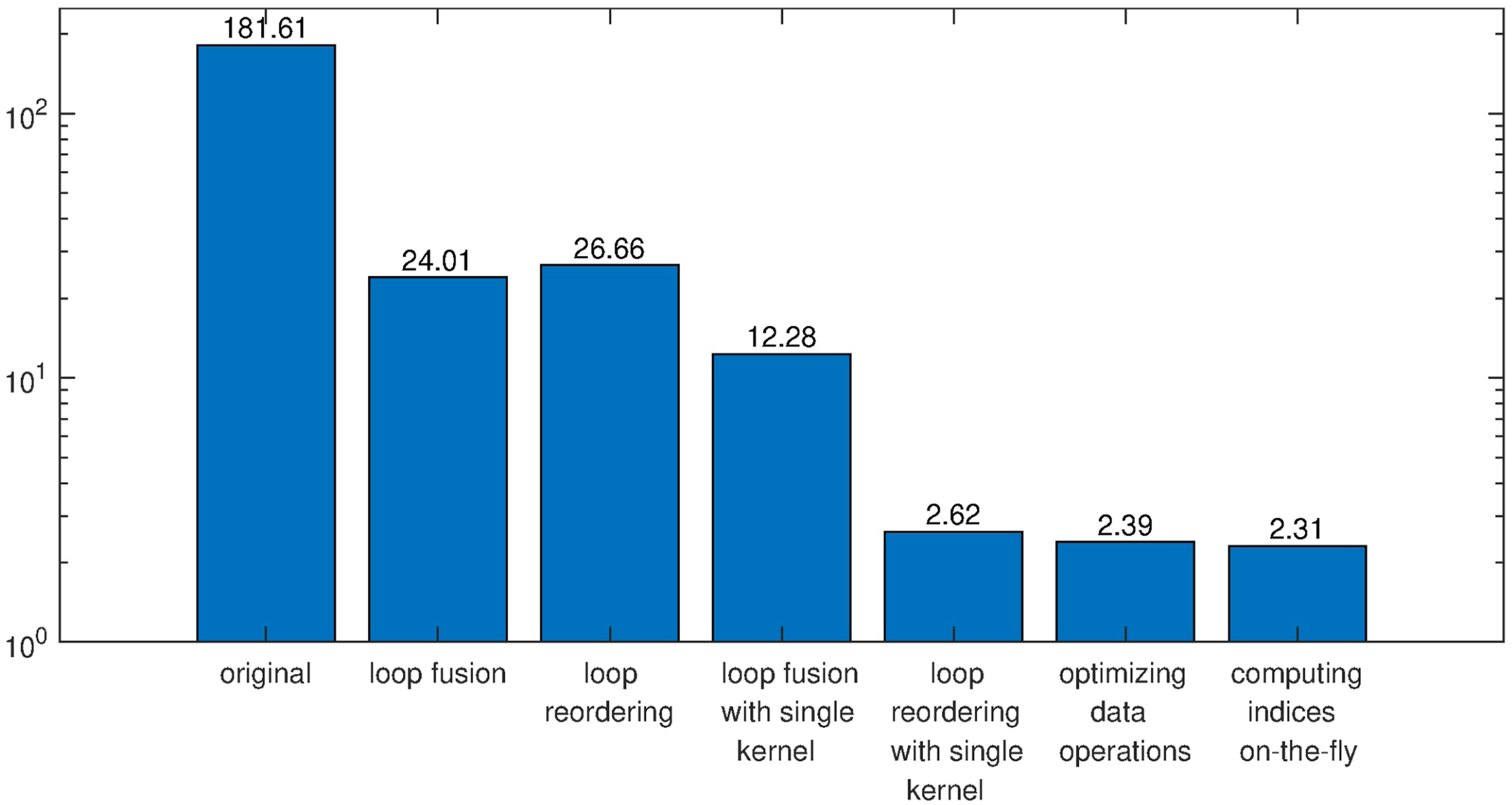
We also used NVIDIA’s Nsight Systems to generate performance profiles of the self-energy calculation before and after optimizations are applied. The Nsight Systems profiles shown in Figures 12 and 13 are generated from a run with n
k
= 1024 and n
t
= 10. We used 16 MPI ranks in the computation, corresponding to 16 A100 NVIDIA GPUs on Perlmutter. Performance profile generated from Nsight Systems for self-energy calculation. The top figure captures the sequence of events occurred in the entire self-energy calculation, while the bottom figure highlights work performed in the first two launched kernels. The profiling result from Nsight Systems for the optimized OpenACC implementation of Σ(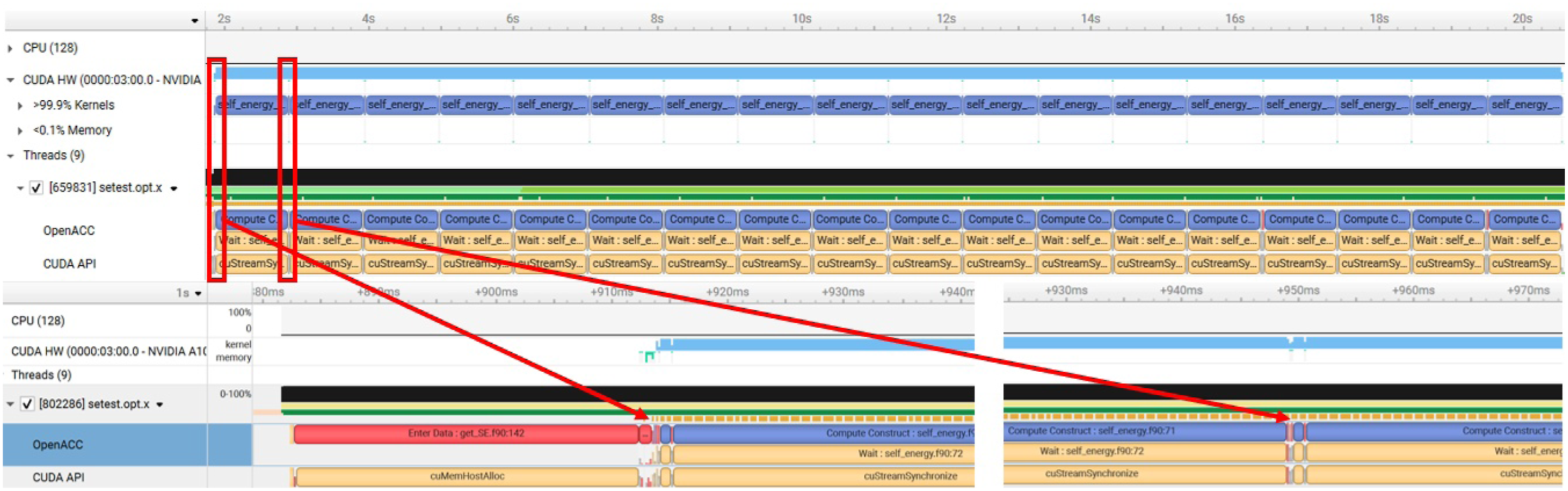
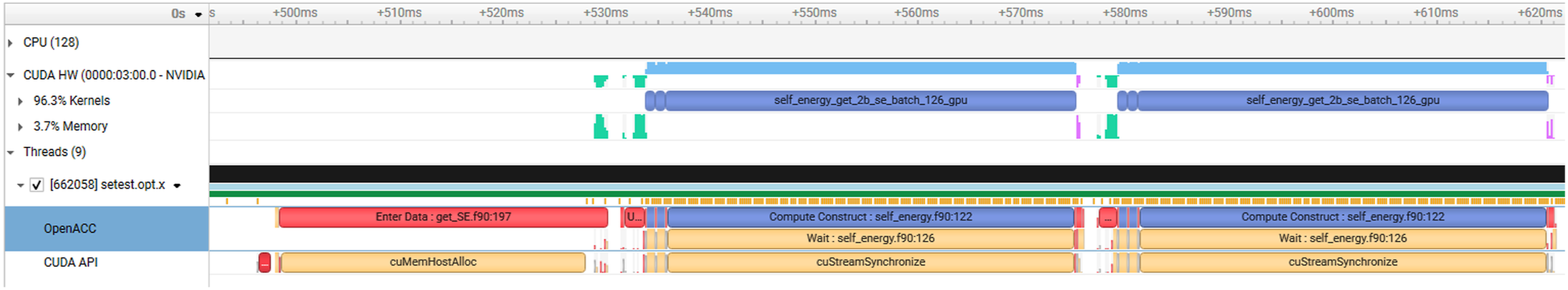
Figure 12 shows the profiling result with direct application of the OpenACC directive
After we apply all the optimizations discussed in the previous subsections, the profiling result becomes what is shown in Figure 13. Compared to Figure 12, there are fewer memory transfers and fewer kernel launches. It only takes about 100 milliseconds for the calculation, which is a significant reduction from 18 seconds used before optimization. For this task, the implementation gives us a reasonable 34% FP64 peak.
5. CUDA FORTRAN implementation
Although OpenACC provides a user friendly programming model to launch concurrent computation on GPU devices, it has some inherent limitations on the management and optimization of GPU resource utilization. Because OpenACC relies on the compiler to partition computational work loads and map them to thread blocks and threads at the compile time, programmers lack complete control over thread utilization and optimization. CUDA FORTRAN NVIDIA Corporation (2023) provides mechanisms for programmers to explicitly define the granularity and hierarchy of concurrent execution, thereby optimizing GPU resource utilization. However, utilizing these mechanisms requires extra effort and meticulous planning. In this section, we show how we use CUDA FORTRAN to implement the self-energy computation. Similar techniques can be used for the collision integral calculations as well.
5.1. Kernel launch
One advantage of the hardware specific CUDA FORTRAN programming model is the direct control it provides over GPUs thread utilization, allowing for customized GPU kernels optimized for performance. A kernel launch requires one to specify the number of thread blocks and the number of threads contained in each block. In CUDA FORTRAN, thread block and thread indices can be organized on a configurable three-dimension grid so that one can easily designate a subset of thread blocks or threads to perform a certain task while other thread blocks or threads can be used to perform other tasks concurrently. We have the flexibility to tailor the grid and block dimensions for each kernel and specify the level of parallelism for individual indices.
Upon analyzing data dependencies, we determine that it is feasible to launch three kernels when calling the subroutine
All the kernels in the CUDA FORTRAN kernel launch for self-energy calculation with thread block and thread configurations specified by 3D arrays 
The dimension of The CUDA FORTRAN implementation of the 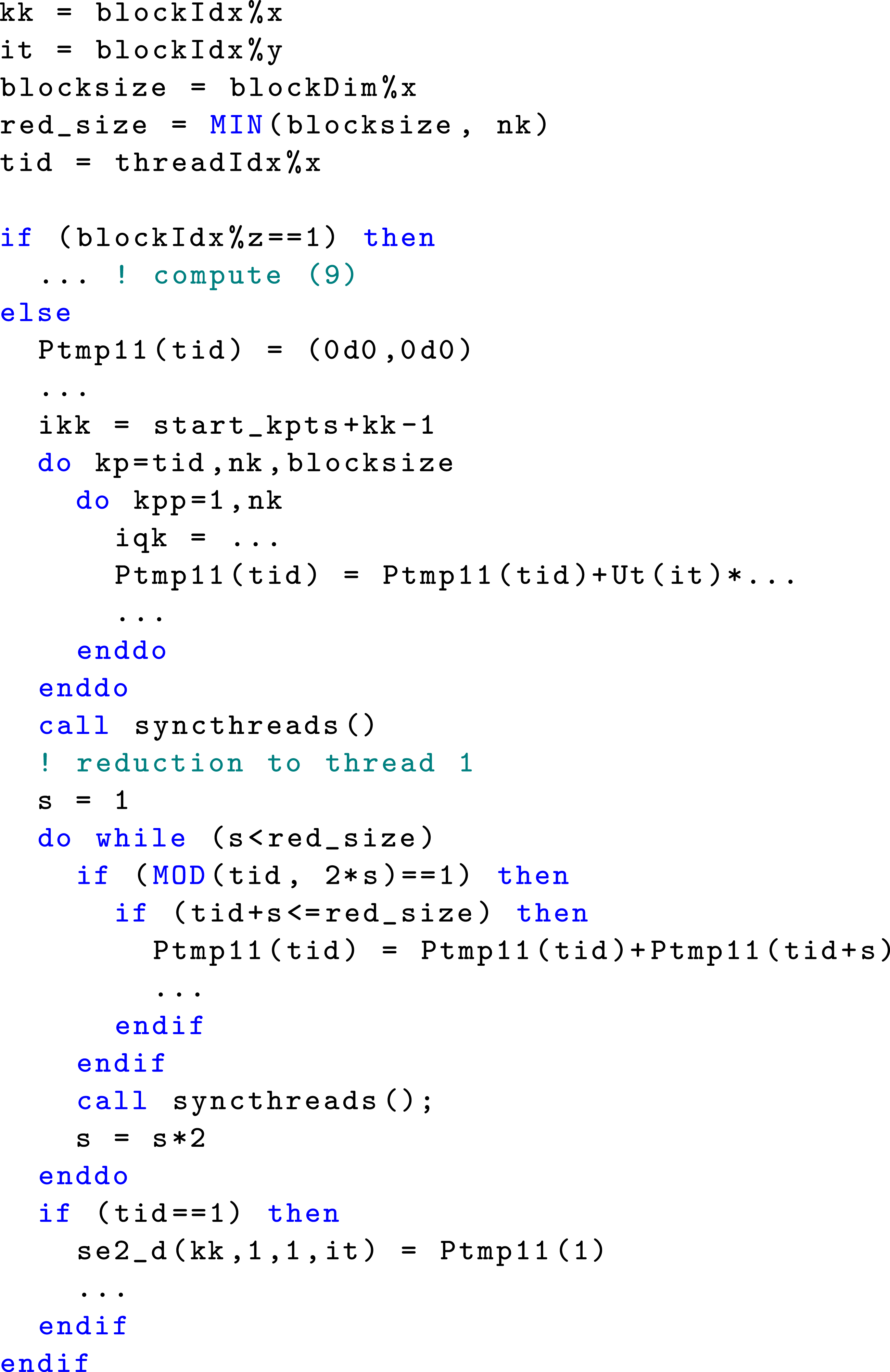
Within the group of thread blocks designated to compute Σ2(
All threads within each thread block are used to execute the nested j, m, q, k′ loops in parallel. The dimension of
5.2. Reduction in the shared memory for summation
As is the case of the OpenMP and OpenACC implementations of the self-energy calculation, a reduction is needed to sum up all terms in
CUDA FORTRAN offers several options to perform the reduction required to sum up several arrays. The easiest option is to use the intrinsic function
It is not clear which reduction algorithm is implemented in the An illustration of a parallel binary-tree based reduction scheme for summing up elements of an array of size 8.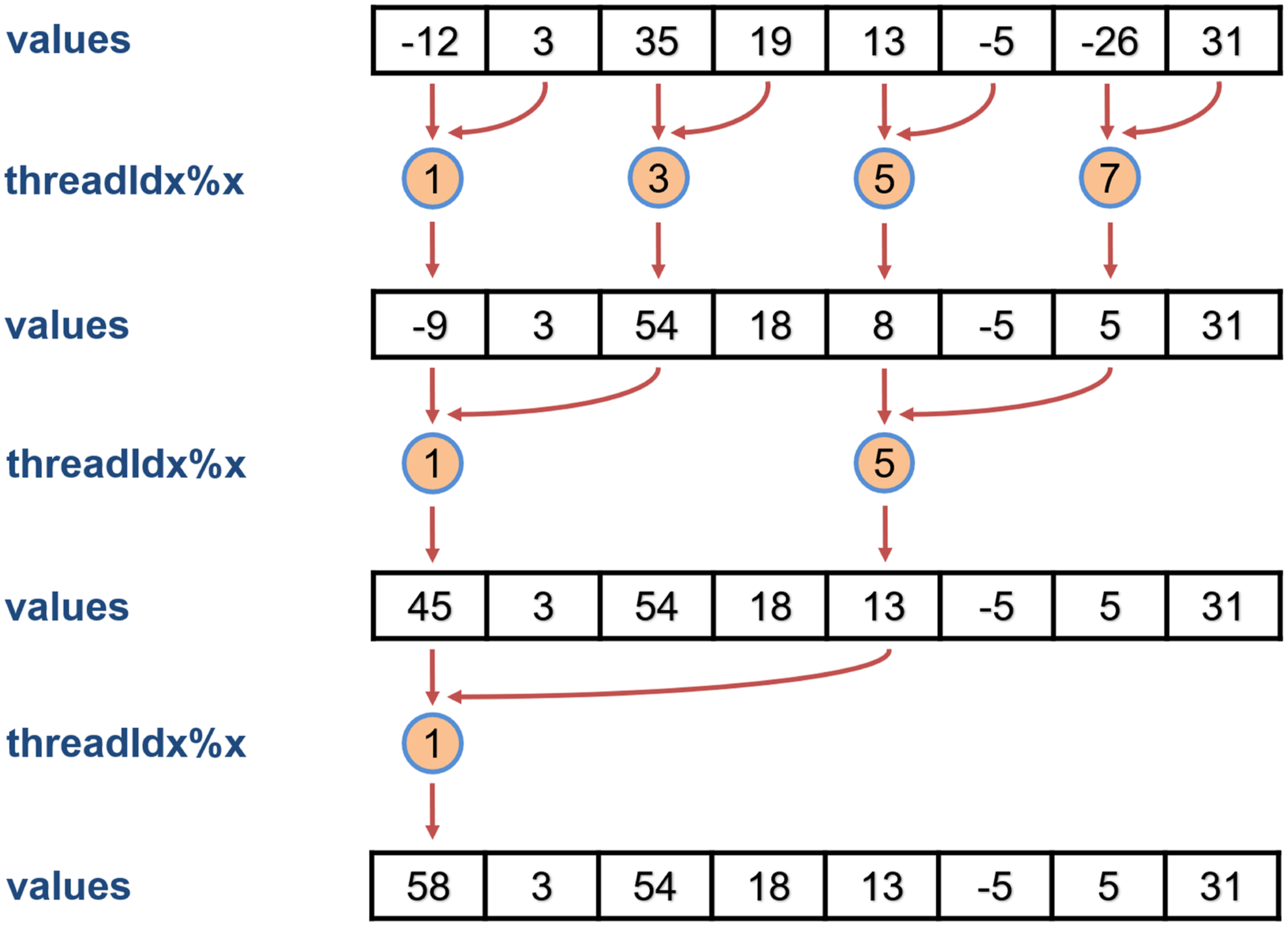
One way to implement this binary-tree based reduction is to use CUDA FORTRAN’s warp-level primitives to repeatedly reshuffle elements of the array to be summed to enable concurrent pairwise summation in a recursive fashion. Figure 17 shows how such a summation is performed for A temporary array 
One drawback of the implementation shown in Figure 17 is that all elements of
Clearly, CUDA FORTRAN provides more flexibility to implement efficient concurrent reduction in shared memory compared to OpenACC which relies completely on the compiler for reduction. Such flexibility results in significant performance improvement.
5.3. Memory management
In CUDA FORTRAN, explicit memory management helps reduce memory allocation and data transfer overheads. Arrays can be directly allocated on the GPU if they are used exclusively on the GPU. Such an array can be passed as an argument to a subroutine and be used on the GPU within the subroutine. This feature eliminates the need to transfer data from a CPU to a GPU and update the data in a coherent fashion, which is required in OpenACC.
By allocating the arrays Device memory allocation and data transfer between host and device in CUDA FORTRAN. In the code snippet, 
CUDA FORTRAN also allows data allocated on the host to be copied to the device and vice versa through a simple assignment statement. For example, in Figure 18, the assignment
is used to copy a portion of the array
While OpenACC does support device-side allocation, managing persistent device-resident data structures and synchronizing them with host-resident MPI buffers can introduce nontrivial overhead.
6. Computational results
6.1. The hardware
We conduct all computational experiments on the Perlmutter supercomputer maintained at National Energy Scientific Computing Center (NERSC). The CPU nodes on Perlmutter has been described in Section 3, and each GPU node on Perlmutter is composed of 1 Milan CPU and 4 NVIDIA A100 (Ampere) GPUs. Each GPU is equipped with 40 GB of high bandwidth memory (HBM) with 1555.2 GB/s GPU memory bandwidth. The peak performance is 9.7 GPU TFlops for the FP64 data type.
6.2. Comparison of the OpenMP, OpenACC and CUDA FORTRAN implementations
In this subsection, we compare the performance of the self-energy as well as the collision integral computation implemented with OpenMP (for CPUs), OpenACC and CUDA FORTRAN (for GPUs). CUDA aware MPI is used to for internode communication.
In the first example, we set n k = 1024, n t = 500 to ensure that a significant amount of work is performed in these calculations and the performance of these calculations can be measured accurately. We then vary the number of MPI ranks used to parallelize the computation among different nodes.
The OpenMP implementation is always run with 64 threads within each MPI rank, 4 MPI ranks are launched on each CPU node.
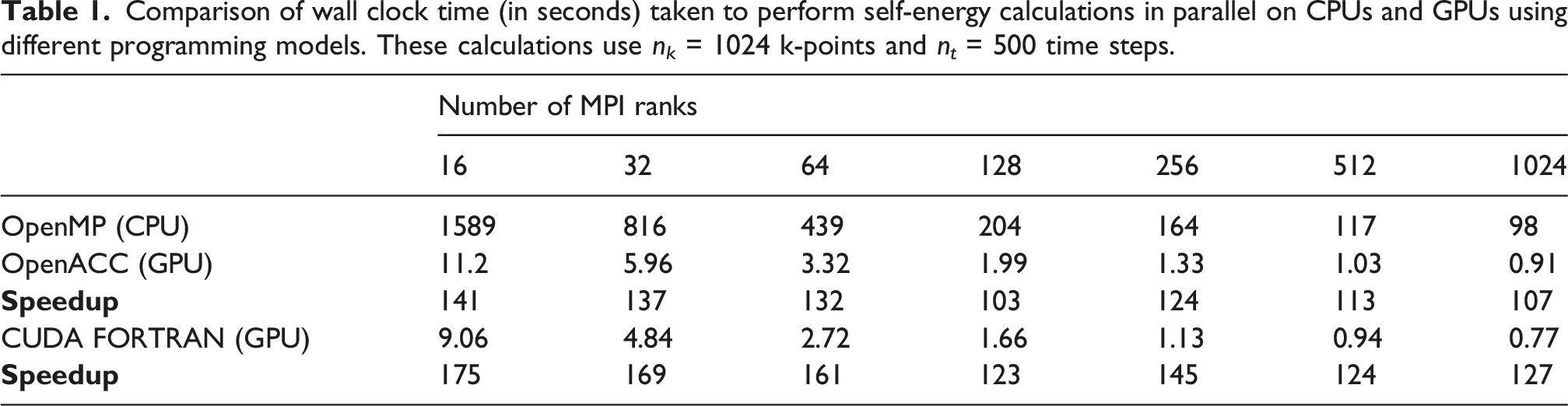
Comparison of wall clock time (in seconds) taken to perform self-energy calculations in parallel on CPUs and GPUs using different programming models. These calculations use n k = 1024 k-points and n t = 500 time steps.
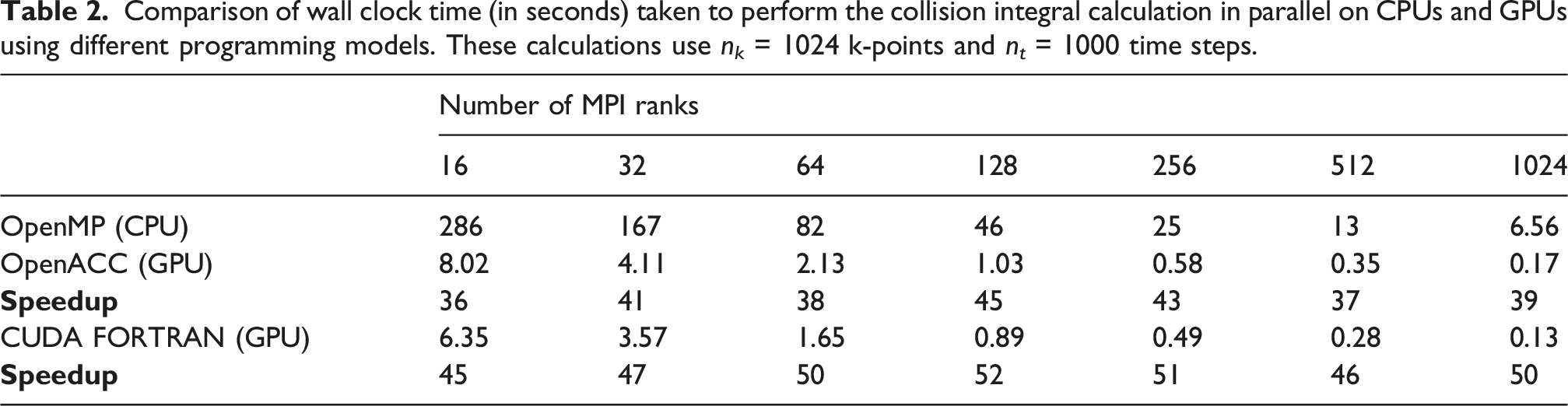
Comparison of wall clock time (in seconds) taken to perform the collision integral calculation in parallel on CPUs and GPUs using different programming models. These calculations use n k = 1024 k-points and n t = 1000 time steps.
In Table 1, we report both the wall clock times used by each implementation to compute the self-energy and the speedup achieved by the OpenACC and CUDA FORTRAN implementations executed on GPUs relative to the OpenMP implementation executed on CPUs. We observe that both the OpenACC and CUDA FORTRAN implementations executed on GPUs are more than 100× faster than the OpenMP implementation executed on CPUs. The CUDA FORTRAN implementation consistently outperforms the OpenACC implementation, which underscores the advantage of having full control over the memory management and kernel launch. On both CPUs and GPUs, the self-energy computation has nearly perfect strong MPI parallel scaling up to 128 MPI ranks. Performance improvement can still be observed with 1024 MPI ranks. However, the scaling beyond 128 MPI ranks is less than perfect. This is due to increased MPI communication overhead relative to reduced computational workload on each MPI rank. Because the computational time consumed on each GPU is significantly lower than that on a CPU, communication overhead becomes more pronounced in GPU runs. As a result, the OpenACC and CUDA FORTRAN speedup over the OpenMP implementation also decreases slightly in general.
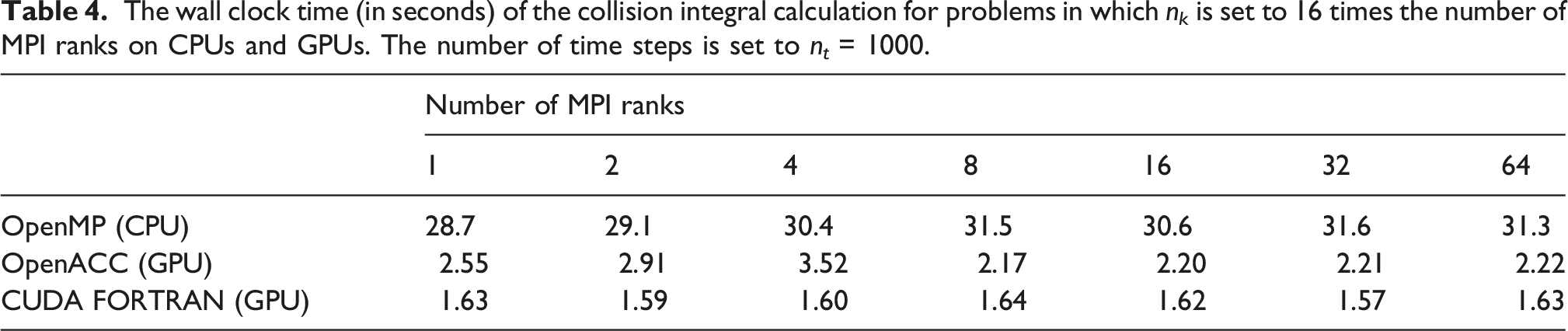
The performance of the collision integral calculation with n t = 1000 is reported in Table 2. We observe that the OpenACC implementation of the collision integral calculation executed on GPUs by OpenACC is consistently 36–45 times faster than the OpenMP implementation executed on CPUs. The CUDA FORTRAN implementation further optimizes the performance, yielding a speedup of approximately 50 times. Furthermore, excellent strong scaling is observed for collision integral calculation. We remark here that because the amount of work in the collision integral calculation is much less than for that in the self-energy calculation, we cannot take full advantage of the concurrency provided by a large number of thread blocks and threads.
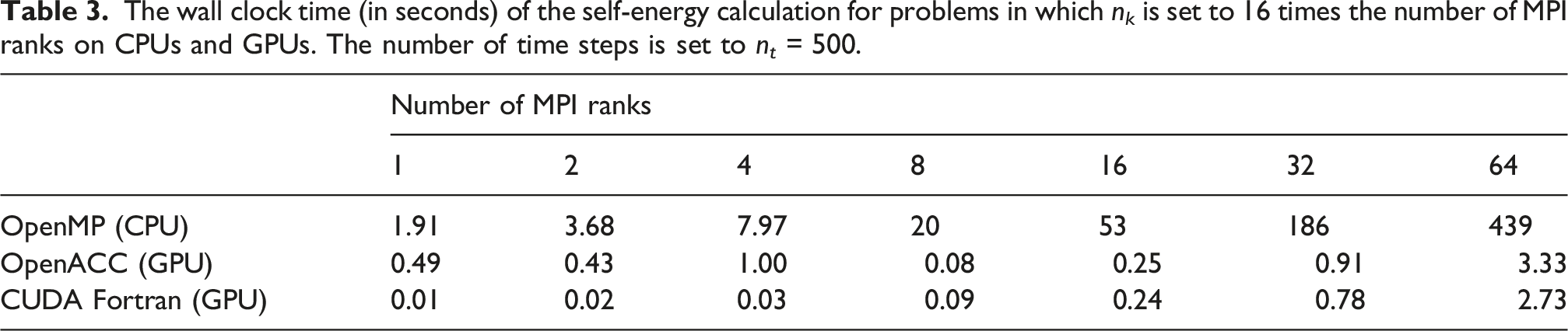
In addition to strong scaling, we also examined the weak scaling of the GPU implementations of the self-energy and collision integral calculation by fixing the number of k-points on each MPI rank to 16. As a result, in each run the total number of k-points which defines the size of the problem to be solved is set to n k = 16× the number of MPI ranks. For this experiment, we keep the values of n t unchanged.
The wall clock time (in seconds) of the self-energy calculation for problems in which n k is set to 16 times the number of MPI ranks on CPUs and GPUs. The number of time steps is set to n t = 500.
The wall clock time (in seconds) of the collision integral calculation for problems in which n k is set to 16 times the number of MPI ranks on CPUs and GPUs. The number of time steps is set to n t = 1000.
6.3. Tuning the grid and block sizes in OpenACC and CUDA FORTRAN
The performance of OpenACC and CUDA FORTRAN implementations varies with respect to several parameters such as the gang and vector size in OpenACC and dimensions of the thread block and thread grids. In this subsection, we examine how different choices of these parameters affect the performance of self-energy calculation. The test results presented below use n
k
= 1024 and n
t
= 500. The computation is distributed among 16 MPI ranks. The wall clock time associated with different choices of vector size and gangle size in OpenACC in shown in Figure 19 as a heatmap. Figure 20 shows the wall clock time of self-energy calculation used by the CUDA FORTRAN implementation for different choices of blockDim and gridDim. These figures illustrate the impact of various gang sizes and vector sizes in OpenACC, as well as different grid and block dimensions in CUDA FORTRAN, respectively. Comparison of wall clock time (in seconds) used in the OpenACC implementation of the self-energy calculation with different gang and vector sizes. These calculations use n
k
= 1024 k-points and n
t
= 500 time steps, and are run on 16 MPI ranks. Comparison of wall clock time (in seconds) used in the CUDA FORTRAN implementation of the self-energy calculation with different thread grid and block sizes. These calculations use n
k
= 1024 k-points and n
t
= 500 time steps, and are run on 16 MPI ranks.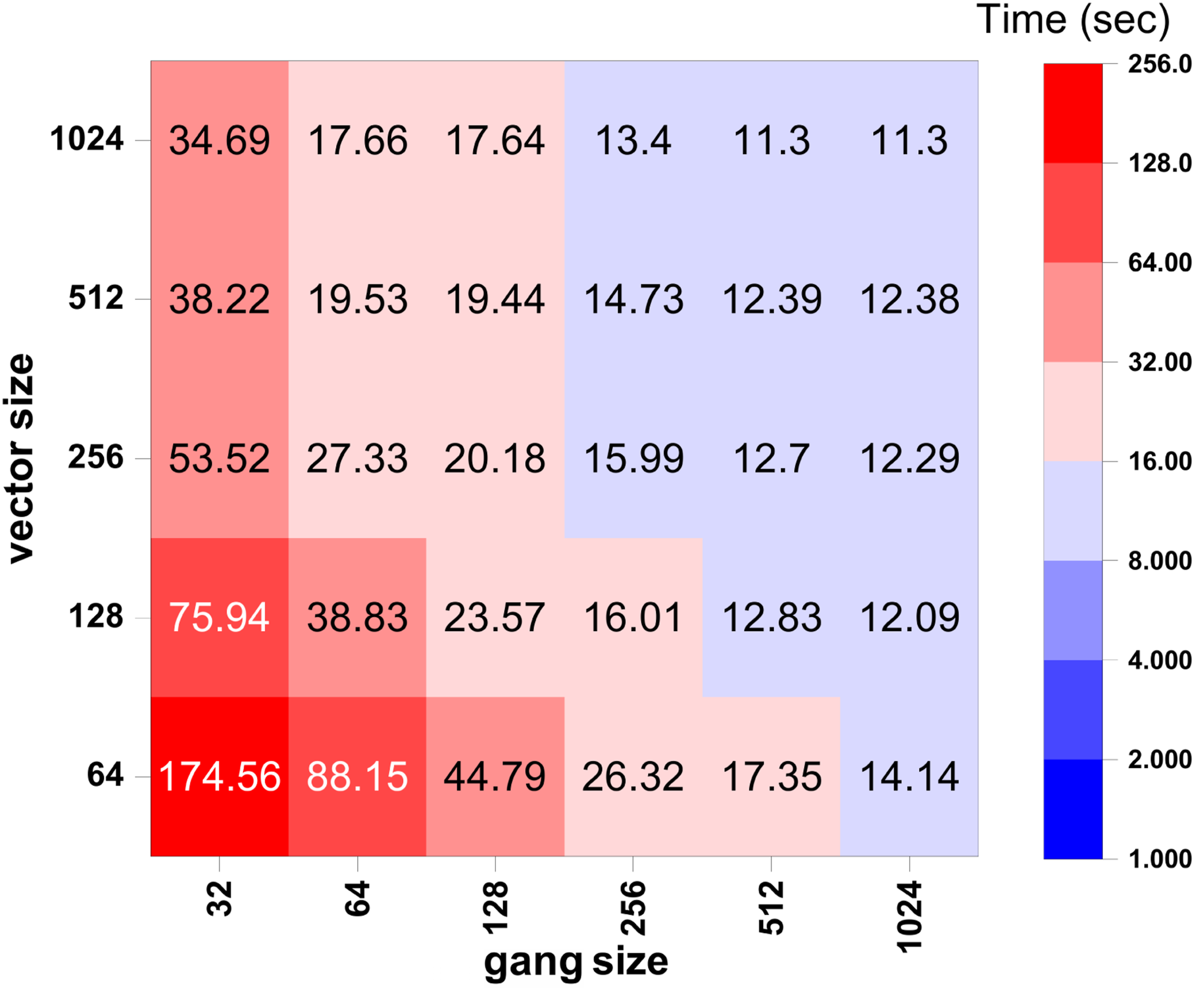
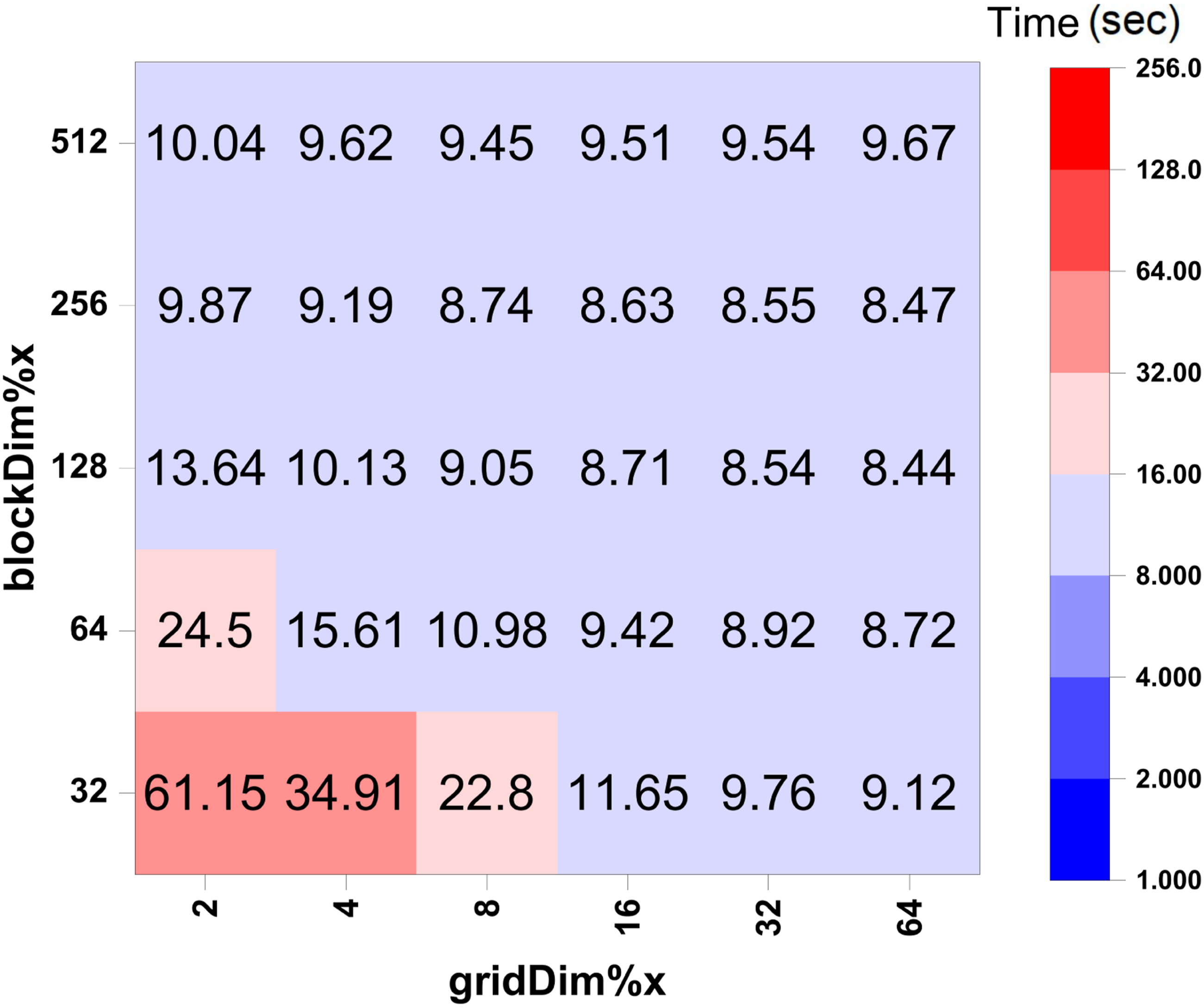
These figures show that in OpenACC, computational time generally decreases as gang and vector sizes increase. A similar trend of reduced computation time with increasing grid and block dimensions is also observed in CUDA Fortran.
In the OpenACC implementation, performance is more sensitive to the choice of gang size. Increasing the gang size to 1024 yields significant improvements, while increasing the vector size from 128 to 512 shows minimal effect. Interestingly, performance improves further with a vector size of 1024. As shown in Figure 19 the optimal configuration for this example appears to be a gang size of 512 and a vector size of 1024.
In the CUDA FORTRAN implementation, the key grid and block dimensions to consider are
7. Related work
Numerical algorithms for solve the KBEs have been described in (Balzer, 2007; Stan et al., 2009; Meirinhos et al., 2022). A parallel implementation of the algorithm for the second-Born self-energy approximation for the Hubbard model was presented in Balzer et al. (2010). Efficient contraction of the second-Born approximation of the self-energy with the Green’s function is discussed in Tuovinen et al. (2019). The focus that work is on contraction on a single processor. A software package called Non-equilibrium System Simulation (NESSi) package Schüler et al. (2020) has recently been made available to the community. NESSi contains a hybrid MPI-OpenMP implementation for CPUs. NESSi uses MPI to parallelize over the k-points, which is the approach we take in this work. OpenMP is used in NESSi to parallelize time-stepping. The parallelization of the self-energy evaluation is left to the user in NESSi. To the best our knowledge, our work is the first to address how to solve KBE efficiently on GPUs. The GPU implementation of collision integrals in the context of electron transport and nonequilibrium dynamics from electron-phonon interactions is recently presented in Peng et al. (2025).
8. Conclusion
In this work, we developed high-performance implementations of a numerical method for solving the Kadanoff-Baym equations (KBE) on both CPU and GPU architectures, using a range of parallel programming models. For GPU-based acceleration, we developed several optimization techniques aimed at improving performance by maximizing computational resource utilization and minimizing overhead associated with kernel launches and memory management.
Our experiments demonstrate that the GPU implementation of the self-energy computation, developed using both OpenACC and CUDA FORTRAN, achieves over 100× speedup on NVIDIA A100 GPUs compared to an AMD EPYC many-core CPU baseline. For the collision integral computation, GPU acceleration yields more than a 10× performance gain. We found that the explicit control over thread hierarchy and memory enabled by CUDA FORTRAN leads to better performance than OpenACC, highlighting the value of fine-grained optimization in GPU programming.
All three implementations (MPI + OpenMP CPU, MPI + OpenACC GPU, and MPI + CUDA FORTRAN GPU) exhibit excellent strong and weak parallel scaling, underscoring the portability and scalability of our approach across modern high-performance computing systems. These results demonstrate the viability of our method for large-scale non-equilibrium Green’s function calculation for quantum many-body systems and contribute to advancing the state of the art in GPU-accelerated scientific computing.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the Center for Computational Study of Excited-State Phenomena in Energy Materials (C2SEPEM) at the Lawrence Berkeley National Laboratory, which is funded by the U. S. Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division, under Contract No. DE-AC02-05CH11231, as part of the Computational Materials Sciences Program. This work is also supported by the U.S. Department of Energy, Office of Science, Office of Advanced Scientific Computing Research, Scientific Discovery through Advanced Computing (SciDAC) program. The authors acknowledge the computational resources of the National Energy Research Scientific Computing (NERSC) center using ASCR-ERCAP-m1027.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.