
Abstract
A woman with Sjögren syndrome manifesting as aphasia with a left deep cerebral white matter lesion tested positive for anti-aquaporin 4 (AQP4) antibody. Open biopsy of the lesion revealed active demyelination with edematous changes and the preservation of most axons, indicating a non-necrotic demyelinating lesion. Immunostaining for AQP4 was diffusely lost, whereas the loss of glial fibrillary acidic protein immunostaining was limited but with highly degenerated astrocytic foot processes in perivascular areas. These results suggested neuromyelitis optica spectrum disorder (NMOSD) pathology rather than Sjögren-related vasculitis. Only cerebral cortical symptoms with a cerebral white matter lesion could be observed in NMOSDs.
Keywords
Introduction
Neuromyelitis optica (NMO) is an inflammatory disease that mainly affects the optic nerve and spinal cord. The anti-aquaporin 4 (AQP4) antibody is specifically found in the sera of patients with NMO. It has also been implicated in various clinical conditions called NMO spectrum disorders (NMOSDs), and has been detected in the sera of Sjögren syndrome (SS) patients with central nervous system (CNS) involvement.1,2 However, the relationship between NMOSDs and SS remains unclear. We present a case of NMOSD complicated by SS with a cerebral white matter lesion and discuss the pathological implications.
Materials and methods
Histochemical and immunohistochemical procedures were performed as previously described. 3
Case report
A 48-year-old woman exhibited arthralgia in her fingers and was diagnosed with rheumatoid arthritis. She presented to a hospital with sudden difficulty in speaking at the age of 50. T2-weighted brain magnetic resonance imaging (MRI) revealed high-signal intensity lesions mainly in the left temporal subcortical white matter. She was referred to our hospital, and a neurological examination revealed disturbances in language comprehension and repetition, hand-finger and constructional apraxia, acalculia, and generalized hyperreflexia of the limbs. Her visual field and acuity were normal. A blood test revealed mild anemia. Thyroid function was normal. Rheumatoid factor, antinuclear antibody, and SS A and B antibodies were positive. Although she was unaware of xerophthalmia or xerostomia, Schirmer’s test and the Rose Bengal test were positive, and a reduction in salivary secretion was confirmed in a gum test, which fulfilled the 1999 Japan Revised SS Criteria. 4 She was diagnosed with SS secondary to rheumatoid arthritis. The cell count and protein concentration in the cerebrospinal fluid (CSF) were normal, and myelin basic protein and oligoclonal bands were negative. Conventional and fluid-attenuated inversion recovery (FLAIR) T2-weighted sequences revealed high-intensity lesions in the subcortical temporal and parietal white matter of the left (Figure 1(A)). On diffusion-weighted image (DWI) the lesions were iso-mild hyperintense and on apparent diffusion coefficient image maps (ADC) hyperintense relative to the normal white matter, excluding the diagnosis of acute infarction. Spinal MRI revealed no abnormalities. An in-house cell-based assay detected the anti-AQP4 antibody in the serum.
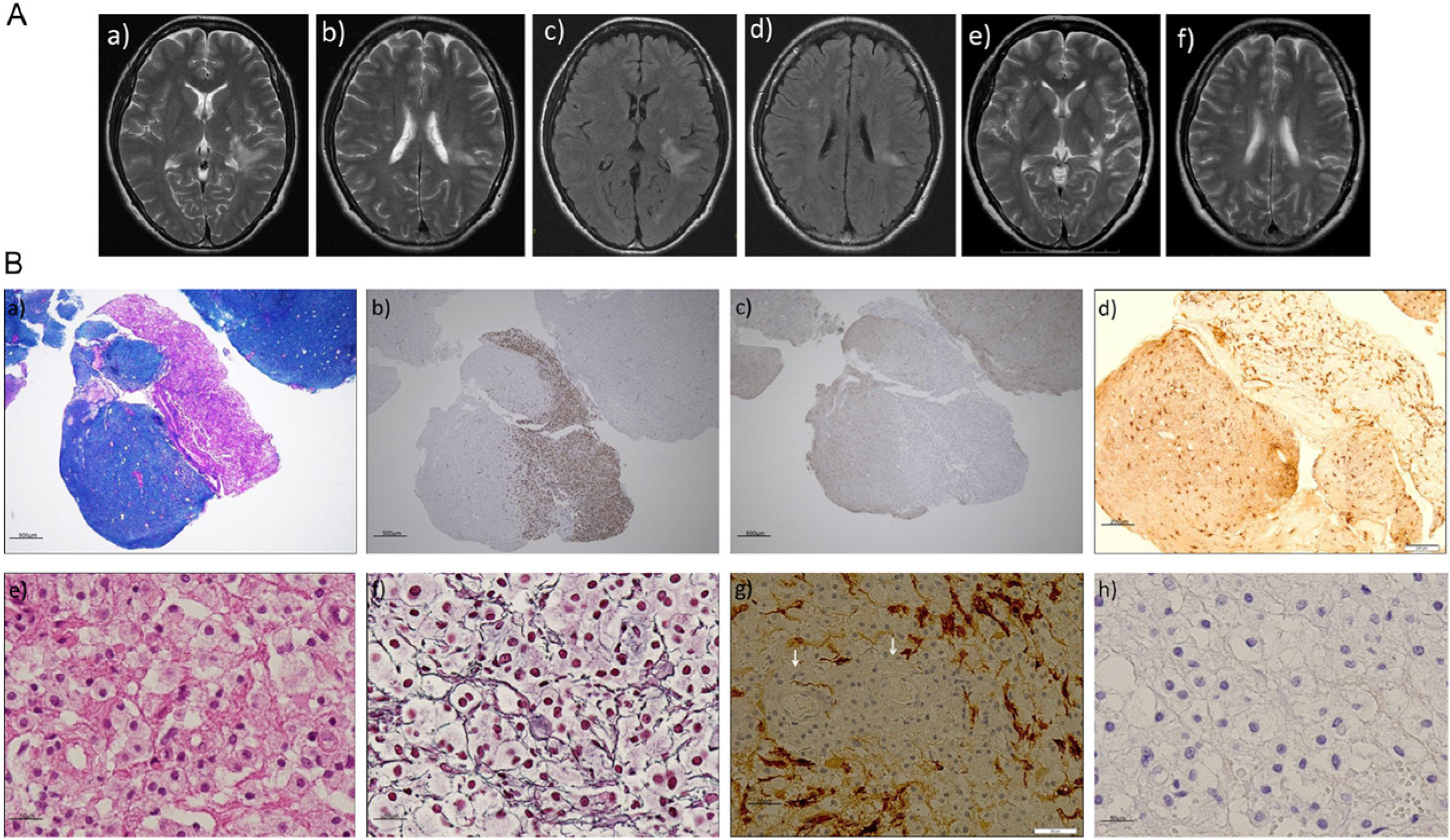
(A) Brain magnetic resonance imaging (MRI) findings.
Open brain biopsy was performed on the white matter in the left temporal lobe. The oral administration of 4 mg/day of betamethasone was initiated after biopsy, and was then tapered gradually. Her symptoms improved to mild constructional apraxia and acalculia, and the white matter lesion on MRI was reduced in size (Figure 1(A)). She currently takes 0.75 mg/day of betamethasone orally, and her symptoms have not deteriorated.
Neuropathological findings (Figure 1(B))
Pathological changes were observed only in the white matter with the infiltration of lipid-laden macrophages and vasogenic edema, suggesting active demyelination (Figure 1(B)-(a), (b)). Immunohistochemical analysis revealed that AQP4 expression was diffusely lost (Figure 1(B)-(c)). Glial fibrillary acidic protein (GFAP) immunoreactivity in the lesion suggested the degeneration of astrocytic foot processes, especially in perivascular areas (Figure 1(B)-(d), (g)).
Massive phagocytic macrophages were observed under high-magnification without vasculitis-related necrotic alterations (Figure 1(B)–(e)), and axons were mostly preserved, even in severely inflamed areas (Figure 1(B)-(f)). The loss of astrocytes and their foot processes was marked in perivascular areas (Figure 1(B)–(g)), and AQP4 was completely lost (Figure 1(B)–(h)).
Discussion
NMO is a severe inflammatory demyelinating disorder characterized by monophasic or relapsing acute myelitis and optic neuritis. Although the lack of cerebral involvement was initially proposed for NMO, accumulating evidence has indicated that cerebral involvement in various regions of the brain, both symptomatic and asymptomatic, is common in NMOSDs. 5 Brain abnormalities were detected on MRI in more than half of NMO patients, which included the cerebral white matter, diencephalon and brainstem.5,6 NMOSDs exhibit various clinical manifestations of brain involvement, including aphasia, hemiparesis and cognitive impairment caused by extensive cerebral white matter lesions.5,6 Therefore, NMOSDs should be considered even if a cerebral white matter lesion, without optic neuritis or spinal cord lesions, is only detected.
Mild astrocytopathy, featuring non-cavitary demyelinating lesions with well-preserved axons, was observed in our case, which differed from the extensive necrosis typically observed in NMO spinal cord lesions. This result may be explained by the pathological diversity of NMO lesions, which includes the typical demyelinating patterns without the loss of astrocytes classified as type 6 in a previous study, a demyelinating lesion with the loss of AQP4, but not GFAP. 7
The neuropathological changes observed in our case were compatible with NMOSDs, although our case was accompanied by SS, which fulfilled the 1999 Japan Revised SS criteria. Various neurological symptoms can occur in patients with SS due to peripheral nervous system and CNS involvement. Cerebral white matter lesions have previously been reported in patients with SS. 8 Min et al. and Estiasari et al. showed that the rates of SS patients with CNS involvement that were positive for the anti-AQP4 antibody were 75% and 31.8%, respectively.1,2 Excluding our patient, only one other case of SS with cerebral white matter lesions that tested positive for the anti-AQP4 antibody has been reported. 2
The pathologies of CNS lesions in SS patients, particularly cerebral white matter lesions, remain unclear. 9 A previous study showed that the CNS pathologies of SS were angiitis and necrosis. 9 The main neuropathological findings of our case were demyelination and mild astrocytopathy, while necrosis and angiitis were not prominent: Thus, the white matter lesion in this case was determined to be more compatible with NMOSD than SS.
The findings of this case suggest a clinical overlap between NMOSDs and SS, and the significant coexistence of anti-AQP4 and anti-SS A/B antibodies. The anti-AQP4 antibody should be assessed in patients with SS affecting the CNS, and pathological verification is also needed in further investigations of NMOSDs and SS with CNS involvement.
Low-dose corticosteroid monotherapy has been recommended empirically to reduce relapses in NMO, 10 as well as CNS involvement in SS. Our patient has continued to take low doses of betamethasone, and has been free from relapses.
The findings of this case are of importance because few studies have examined pathological changes in CNS lesions in NMOSD patients with SS who tested positive for the anti-AQP4 antibody. Although the neuropathological findings in our patient implicated astrocytopathy in the cerebral white matter lesion in NMOSD with SS, further studies on the relationship between cerebral lesions in NMOSDs and SS are warranted.
Footnotes
Conflict of interest
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.