
Abstract
Background:
Myelitis accompanied by a negative spinal cord MRI may lead to diagnostic uncertainty.
Objective and Methods:
We retrospectively investigated the frequency of negative spinal cord MRI (performed <6 weeks from onset) in Mayo Clinic patients with myelin oligodendrocyte glycoprotein (MOG)-IgG-associated myelitis (2000–2019).
Results:
The initial spinal cord MRI was negative in 7/73 (10%) patients, despite severe acute disability (median EDSS, 7 (range, 4.5–8)); myelitis symptoms/signs were frequent (paraparesis, neurogenic bladder, sensory level, Lhermitte’s phenomenon). Myelitis lesions became overt at follow-up MRI in three patients.
Conclusions:
A negative spinal cord MRI should not dissuade from MOG-IgG testing in patients with acute/subacute myelitis.
Keywords
Introduction
Myelitis is a common manifestation of myelin oligodendrocyte glycoprotein (MOG)-IgG-associated disorder (MOGAD) that typically results in acute severe disability, although recovery is often favorable with prompt immunotherapy. 1 Spinal cord MRI may show longitudinally extensive T2-hyperintense lesions (⩾3 vertebral-body segments), “short” lesions, or both, which are sometimes gray matter–restricted in a linear configuration on sagittal images and/or “H”-shaped axially. 1 However, MOG-IgG-associated myelitis with negative spinal cord MRI has been reported, which could lead to diagnostic uncertainty and treatment delay.2,3 In this paper, we investigated the frequency and characteristics of patients with MRI-negative myelitis associated with MOG-IgG.
Methods
Patients
We retrospectively identified MOGAD patients seen at Mayo Clinic (1 January 2000–31 August 2019) with (1) first clinical myelitis, (2) spine MRI obtained acutely (⩽6 weeks from myelitis onset), and (3) no documented spinal cord T2-abnormalities on radiology reports. MOG-IgG positivity was confirmed by fluorescence-activated-cell-sorting live cell–based assay, as previously reported. 1 We excluded four patients with unavailable MRI and one patient with concomitant MRI brainstem abnormalities. Forty-nine patients were previously reported. 1
MRI-negative myelitis
Two neurologists (E.S.; E.P.F.) reviewed medical records to confirm definitive myelopathic symptoms/signs in MRI-negative patients; consensus was reached after discussion in case of disagreement.
Statistics
Wilcoxon-rank-sum and Fisher’s exact tests were used for comparisons as appropriate (JMP Pro 14.1.0).
Results
Among 73 MOGAD patients at first myelitis episode, the initial spinal cord MRI was interpreted as normal in 7 (10%). When comparing these patients to those with abnormal MRI, there was no statistically significant difference in the percentage of patients treated with immunotherapy prior to MRI between the two groups (2/7 (29%) vs 10/65 (15%) p = 0.3), while the interval (days) from myelitis onset to the first spine MRI was longer in those with normal MRI (median (range), 10 (5–30) vs 4.5 (0–28); p = 0.01).
Patients’ characteristics
The clinical presentations of the seven patients with MRI-negative myelitis are summarized in Table 1. All tested negative for aquaporin-4-IgG.
Clinical presentations in seven patients with MRI-negative myelitis.

MOGAD: myelin oligodendrocyte glycoprotein (MOG)-IgG-associated disorder; CSF: cerebrospinal fluid; EDSS: expanded disability status scale–score; EMG/NCS: electromyography/nerve conduction study; IVIG: intravenous immune globulin; PLEX: plasma exchange.
Median age at myelitis was 45 years (range, 35–80), 3 (43%) were female, and all were Caucasian. The myelitis occurred at MOGAD presentation in all. Myelitis symptoms/signs included the following: sensory disturbances, 7; bowel/bladder dysfunction, 7 (requiring catheterization in 5); weakness, 6; upper motor neuron signs (spasticity, hyperreflexia, or positive Babinski), 4; trunk sensory level, 3; and Lhermitte’s phenomenon, 3. One patient had an isolated myelitis while six had other neurologic manifestations (optic neuritis, 4; headache, 4; encephalopathy, 2). Three had viral-like prodromes. Median expanded disability status scale (EDSS)–score at nadir was 7 (range, 4.5–8). Median MOG-IgG titer was 100 (range, 100–10,000). Cerebrospinal fluid analysis, available in six, revealed pleocytosis (5/6; median white cell count, 105/µL (range, 28–204)) and absence of oligoclonal bands (6/6). Somatosensory-evoked potentials were obtained in two patients revealing conduction delay localizable to the spinal cord in both. Two patients had normal nerve conduction/electromyography while one had conduction block and denervation changes concomitant with a spastic paraparesis confirming a myelo-neuropathy.
On MRI re-review (E.S.; K.N.K.; E.P.F.), one patient showed a subtle focal area of T2-hyperintensity on axial image (Figure 1(a1–a4)). Six patients underwent spine MRI with gadolinium, but none had abnormal enhancement. Myelitis lesions became overt in three of six patients who underwent follow-up MRI after a median of 6 days (range, 6–26; Figure 1(d1–d8)). Brain MRI, available in five, showed concomitant brain parenchymal abnormalities in one (Figure 1(e1–e2)).
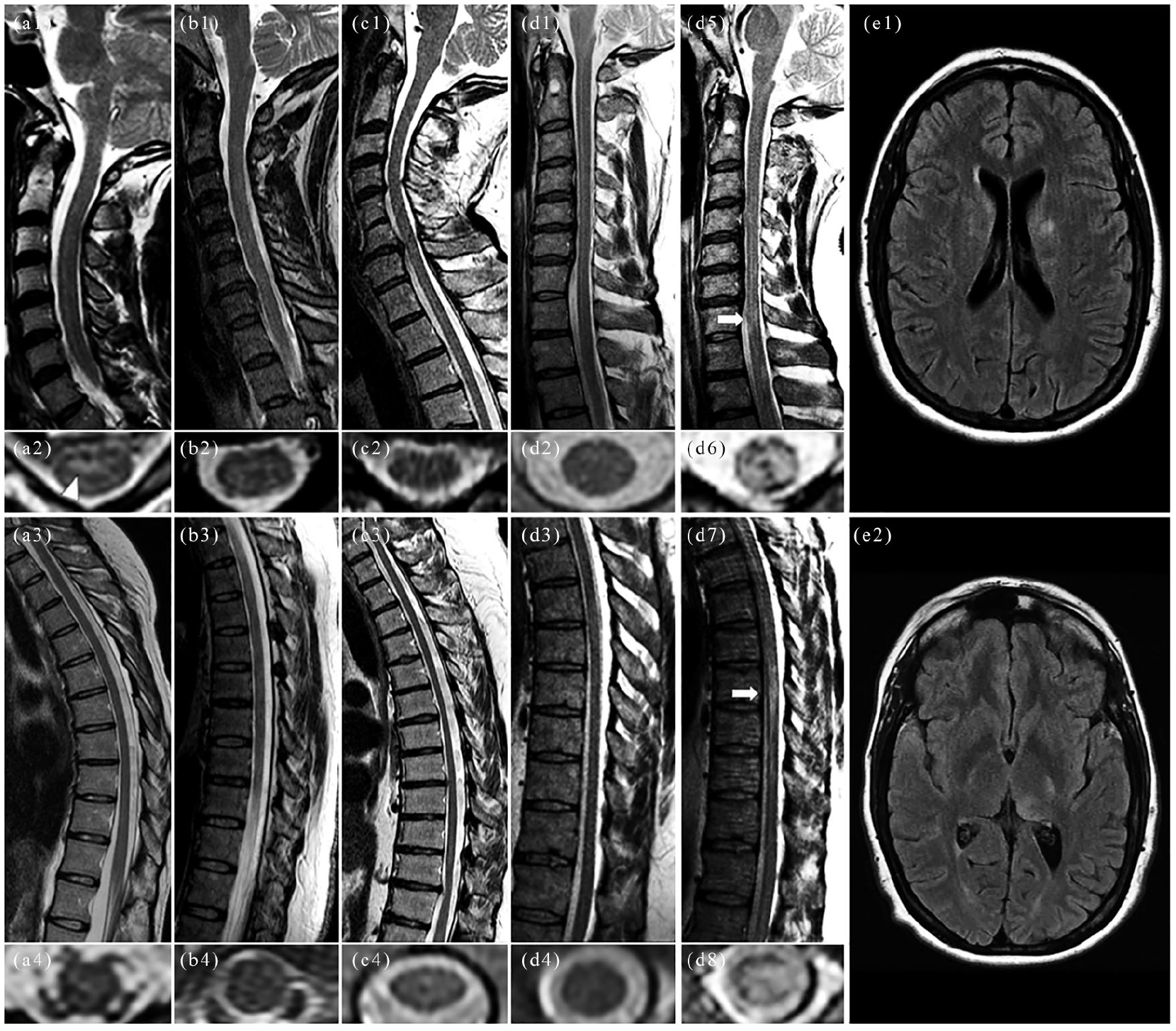
Representative examples of negative spinal cord MRI in patients with MOG-IgG-associated myelitis.
All patients received acute immunotherapy (corticosteroids, 7; intravenous immunoglobulin, 2; plasmapheresis, 2) with improvement. Alternative diagnoses initially assigned in the setting of MRI-negative myelitis included the following: bacterial/viral encephalitis, 2; Guillain-Barre syndrome, 1; and amyotrophic lateral sclerosis, 1. Four patients had typical MOGAD relapses including optic neuritis, encephalitis, and myelitis.
Discussion
In this study of MOGAD patients at the time of their first episode of clinical myelitis, the initial spinal cord MRI was negative in 10% of cases, despite severe myelitis manifestations. A negative acute MRI should not dissuade from MOG-IgG testing with a subacute myelopathy.
A prior study reported a negative MRI in 1 of 28 patients (4%) with MOG-IgG-associated myelitis, not focusing on the first myelitis episode. 4 Although typically not expected with other well-defined acute/subacute myelitis (e.g. aquaporin-4-IgG-associated myelitis), an initially negative MRI may occur in idiopathic transverse myelitis (5%), 5 acute flaccid myelitis (5%), 6 and paraneoplastic myelopathies (35%). 7 It is also seen with glycine receptor and glutamic acid decarboxylase-65 antibodies, but the exact frequency is unknown. 8 Faint T2-hyperintensity is typical of glial fibrillary acidic protein (GFAP)-IgG-associated myelitis and generally has accompanying brain involvement (e.g. radial perivascular enhancement). 9 Finally, an initially negative MRI is seen in up to 24% of cases of spinal cord infarction. 10 Spinal cord infarction would usually have a more hyperacute presentation (nadir < 4–12 hours), absence of cerebrospinal fluid pleocytosis, lack of concomitant/preceding brain/optic nerve involvement, and no viral-like prodrome.1,10
The reason why some patients with MOGAD myelitis have negative MRI is uncertain but could relate to imaging timing (late imaging missing a transient lesion or early imaging missing an evolving lesion), 11 insensitivity of MRI to the detection of MOG-IgG-associated inflammation, a spinal meningitis without myelitis, or a functional disturbance mediated by MOG-IgG binding. The longer time to MRI in the imaging-negative group could be explained by those patients having a more slowly evolving clinical course or statistical chance in the setting of small numbers. Patients with MOGAD myelitis may show questionable T2-hyperintensities on MRI that are in contrast to the severity of myelitis symptoms or can be interpreted as physiological variants (e.g. a faint linear T2-hyperintense on sagittal images may be difficult to discern from a prominent central canal). In uncertain cases, careful evaluation of axial images or repeat MRI after days/weeks may confirm the myelitis (Figure 1). Somatosensory-evoked potentials may also help confirm the spinal cord localization, further justifying MOG-IgG testing. The abnormal electromyographic findings in one patient coexisting with a spastic paraparesis suggested a myeloradiculitis which is recognized to occur with MOGAD. 12
Our study is limited by the retrospective nature and potential referral bias (atypical cases might be over-represented at our tertiary care center). One patient had clinical/MRI-evidence of brain involvement that could have contributed to their symptoms. However, the constellation of clinical findings (e.g. urinary retention, lower limb numbness, weakness, and spasticity/hyperreflexia) and sparing of the brainstem on brain MRI strongly supported a myelopathy.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: S.J.P. reports grants, personal fees, non-financial support, and other from Alexion Pharmaceuticals, Inc.; grants from Grifols; other from Euroimmun; grants from National Institutes of Health (NIH); grants, personal fees, and non-financial support from Guthy-Jackson Charitable Foundation; grants from AEA (Autoimmune Encephalitis Alliance); and grants, personal fees, non-financial support, and other from MedImmune, Inc.; in addition, he has a patent (patent #8889102) (application#12-678350) on neuromyelitis optica autoantibodies as a marker for neoplasia, and also a patent (patent #9891219B2) (application#12-573942) on methods for treating neuromyelitis optica (NMO) by administration of eculizumab to an individual that is aquaporin-4 (AQP4)-IgG autoantibody positive; patents on GFAP-IgG, Septin-5-IgG, MAP1B-IgG, Kelch-like protein 11, and PDE10A are all pending. B.G.W. receives royalties from RSR Ltd, Oxford University, Hospices Civil de Lyon, and MVZ Labor PD Dr. Volkmann und Kollegen GbR for a patent of NMO-IgG as a diagnostic test for NMO and related disorders. He serves as a member of an adjudication committee for clinical trials in NMO being conducted by VielaBio and Alexion pharmaceutical companies. He is a consultant for Chugai Pharma and Mitsubishi Tanabe regarding potential clinical trials for NMO. E.P.F. receives research support as a site principal investigator in a randomized placebo-controlled clinical trial of inebilizumab (a CD19 inhibitor) in neuromyelitis optica spectrum disorders funded by MedImmune/Viela Bio. E.S., K.N.K., D.D., A.S.L., A.K., and N.L.Z. have nothing to disclose.
Ethical Approval
The study was approved by the Institutional Review Board of the Mayo Clinic.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We would like to acknowledge funding support from the NIH National Institute of Neurological Disorders and Stroke (R01NS113828). This study was also supported by the Guthy-Jackson Charitable Foundation and the Mayo Clinic Center for Multiple Sclerosis and Autoimmune Neurology that provided support through a translational clinical fellowship grant.
Informed Consent
All patients consented to the use of their medical records for research.