
Abstract
Background
Inflammation is implicated in the pathogenesis of dementia. However, its role in the vascular cognitive burden and the clinical stage of Alzheimer's disease (AD) remains controversial.
Objective
This study aimed to explore the association of plasma inflammatory proteins with vascular cognitive burden and clinical stage of AD by using a multi-method approach.
Methods
We included 330 individuals with complete plasma protein profiles, Hachinski Ischemia Scale scores, and cognitive function status between September 13, 2005, and October 24, 2007. We employed generalized linear models, restricted cubic splines, and the inverse variance weighting (IVW) method for two-sample Mendelian randomization (MR) to investigate the correlation between inflammatory biomarkers and dementia. We employed the random forest algorithm to build predictive models and utilized SHapley Additive exPlanations (SHAP) analysis for feature importance and interpretability.
Results
AD clinical stage exhibited significant associations with cortisol, C-peptide, tumor necrosis factor receptor-2 (TNFR-2) and interleukin-16 (IL-16, all p < 0.05). Similarly, the vascular cognitive burden was significantly correlated with C-peptide, carcinoembryonic antigen and TNFR-2 (all p < 0.05). These observational findings were corroborated by SHAP analysis. Subsequent MR analysis further revealed a weak negative causal relationship between AD and IL-16 (pIVW = 0.003; ORIVW = 0.981; 95% CI: 0.969–0.994).
Conclusions
Our study identified several inflammatory proteins correlated with the vascular cognitive burden and AD clinical stage, providing exploratory evidence for future mechanistic and interventional research.
Keywords
Introduction
Dementia is among the leading causes of death, especially in the elderly population. 1 Due to the aging population, its prevalence is projected to double every 20 years globally. 2 In many regions, Alzheimer's disease (AD) and vascular dementia (VaD) are among the most common dementia subtypes, each with distinct pathophysiological mechanisms. 3 AD is a progressive neurodegenerative disorder characterized by cognitive decline, memory impairment, and behavioral changes attributed to amyloid-β (Aβ) plaques, abnormal tau tangles, as well as inflammation. 4 VaD is associated with vascular brain injury and fluctuating cognitive symptoms resulting from oxidative stress and inflammation. 5 Similar to AD, the incidence of VaD increases steeply with age, contributing to the growing epidemic of dementia worldwide and imposing a heavy burden on the public health care systems.6,7 This highlights the urgent need for effective biomarkers to facilitate early diagnosis and guide targeted therapeutic interventions. 8
Extensive studies implicate inflammation as a key event in the pathogenesis of dementia. 9 Cytokines and chemokines in neuroinflammation can lead to the development of AD, while risk factors for vascular cognitive impairment can cause inflammation and vascular oxidative stress, which in turn contribute to the vascular cognitive burden.10,11 Many studies support the important role of inflammation in dementia, while others highlight the complexity of this association, particularly in the case of AD. 9 For example, a clinical trial on anti-inflammatory drugs therapy implied that the role of inflammation in AD might not align with our initial expectations. 12 Another study has also revealed that the relationship between inflammation and AD may vary among individuals. 13 These findings indicate that although inflammation plays a critical role in the pathology of dementia, its specific effects on the clinical stage and development of dementia are yet to be fully elucidated.
Hachinski Ischemia Scale (HIS) is a widely used clinical tool designed to evaluates potential vascular contributions to cognitive impairment by examining relevant clinical indicators such as abrupt onset and a history of stroke. 14 The HIS has proven to be the simplest, most sensitive, and most widely used instrument for identifying a cerebrovascular component of cognitive impairment or dementia.15,16 Although the HIS is now less commonly employed than in earlier decades, it can still help identify and discuss key clinical features that distinguish multi-infarct from primary degenerative dementia. 17 Moreover, a recent study has further underscored the effect of HIS as a valuable tool in the research of VaD. 18 However, current understanding on the association between plasma inflammatory proteins and HIS scores remains limited. Investigating the relationship between them could elucidate the association between systemic inflammation and vascular cognitive burden, offering insights into the vascular components of cognitive impairment.
However, observational studies often face biases due to confounding factors or reverse causality, limiting their ability to robustly identify causal associations. 19 To address these limitations, Mendelian randomization (MR) has recently emerged as an effective approach to helping establish potential causal relationships between exposures and diseases.19–21 However, despite extensive studies on inflammatory biomarkers of AD, only a few have explored the causal relationship between genes related to these proteins and AD. Although several studies have investigated the relationships between AD and polymorphisms in tumor necrosis factor-α (TNF-α), TNF receptor-2 (TNFR-2), interleukin-16 (IL-16), and IL-18, none of them have performed MR analysis to further explore these relationships.22–25
To clarify whether inflammation-related plasma proteins track clinical stage across the AD spectrum and vascular burden, we integrated multivariable regression, restricted cubic splines for dose-response characterization, two-sample MR for causal triangulation where valid instruments were available, and explainable machine learning to assess predictive contributions. The objective of this study was to investigate the associations between plasma inflammatory proteins and both the vascular cognitive burden and AD clinical stage. Our goal was to identify plasma inflammatory biomarkers that can predict vascular cognitive burden and AD clinical stage independently, and to validate the biomarkers through multiple analytical approaches.
Methods
Study population
Data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The original goal of ADNI was to test whether serial magnetic resonance imaging, positron emission tomography, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early AD. The current goals include validating biomarkers for clinical trials, improving the generalizability of ADNI data by increasing diversity in the participant cohort, and to provide data concerning the diagnosis and progression of AD to the scientific community. For up-to-date information, see adni.loni.usc.edu.
Although ADNI was designed to investigate the continuum of AD rather than VaD, it remains one of the largest publicly available repositories with concurrent plasma proteomics and detailed clinical phenotyping. However, only the ADNI-1 cohort, which was recruited between September 13, 2005 and October 24, 2007, includes the full panel of plasma proteins. As a result, a total of 566 participants with complete data on plasma protein levels, HIS scores and cognitive function status in the ADNI database were included in this study. Participants who met the following criteria were excluded from this study: 1) individuals with missing information on gender, age, or Apolipoprotein E (APOE) genotype, 2) individuals with a history of long-term use of antibiotics or immunomodulatory drugs, and 3) individuals with neurological histories other than dementia. Finally, 330 participants were included into the study. The present work comprises: (i) an observational association analysis of plasma inflammatory proteins with cognitive diagnosis and HIS at baseline, and (ii) an external two-sample MR analysis using publicly available Genome-wide association study (GWAS) summary statistics to assess potential causality.
Plasma protein measures
Plasma proteins were quantified using the Biomarkers Consortium ADNI Plasma Targeted Proteomics Project, which measured 190 analytes on the Luminex xMAP platform. Plasma protein concentrations were log10-transformed to reduce right-skewness and approximate normality. All analyses involving proteins were conducted on the transformed scale; therefore, summary statistics and model coefficients should be interpreted per 1-unit increase in log10 concentration. All protein variables were standardized to z-scores prior to regression to facilitate comparability across biomarkers.
Outcome measures
The HIS, one of the most commonly used clinical tools for quantifying the vascular cognitive burden, was employed in this study. 26 HIS scores were calculated based on the following criteria: sudden onset (2 points), stepwise deterioration (1 point), fluctuating course (2 points), nocturnal confusion (1 point), relative preservation of personality (1 point), depression (1 point), somatic symptoms (1 point), emotional incontinence (1 point), history of hypertension (1 point), history of stroke (2 points), atherosclerosis (1 point), focal neurological symptoms (2 points), and focal neurological signs (2 points). 17 Higher scores denote greater vascular cognitive burden: a HIS ≥ 7 indicates prominent vascular cognitive burden, ≤ 4 implies minimal vascular cognitive burden, and 5–6 reflects intermediate vascular cognitive burden. 14
The AD clinical stage was classified into three categories based on participants’ cognitive function status: normal level (NL), MCI, and dementia. NL participants had no memory complaints, a Clinical Dementia Rating (CDR) score of 0, and a Mini-Mental State Examination (MMSE) score of 24–30. MCI participants had memory complaints verified by study partners, intact activities of daily living, a CDR score of 0.5, and an MMSE score of 24–30. Dementia was diagnosed in participants who met the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer's Disease and Related Disorders Association criteria for probable AD, and had memory complaints, a CDR score of 0.5 or 1.0, as well as an MMSE score of 20–26. More details can be found on the ADNI website (https://adni.loni.usc.edu).
Statistical analysis
We summarized missingness for all exposures, outcomes, and covariates. The primary analyses were conducted using a complete-case approach. Normality was assessed using the Shapiro-Wilk test. Normally distributed variables are presented as mean ± SD and analyzed by one-way ANOVA; skewed variables are presented as median (IQR) and analyzed using non-parametric tests. Categorical variables were analyzed using chi-square tests or Fisher's exact test when expected counts < 5, and were presented as percentages. Logistic regression models were used to evaluate the associations between inflammatory proteins and AD clinical stage, while generalized linear models were utilized to assess the associations between inflammatory proteins and HIS scores, adjusting for gender, age, education level, and APOE genotype. Covariates were selected a priori based on clinical relevance and prior literature. Multiple comparisons were corrected using the false discovery rate (FDR) approach. Observed associations were rigorously validated by 2000 bootstrap resamples. To further explore the associations between inflammatory proteins and both AD and HIS scores, restricted cubic splines (RCS) were employed to investigate potential non-linear relationships between them. Restricted cubic splines with 4 knots placed at the 5th, 35th, 65th, and 95th percentiles were used. Nonlinearity was tested using a likelihood ratio test comparing the spline model to a linear term.
Additionally, two-sample MR was conducted to explore the causal relationships between plasma inflammatory proteins and AD. Data from GWAS were used to select single nucleotide polymorphisms (SNPs) serving as instrumental variables (IVs) for MR analysis. The analysis basically relied on three assumptions: (1) the selected IVs were strongly associated with the exposure, (2) independent of confounders, and (3) only affected the outcome through the exposure. To ensure robustness, we set the significance threshold for IVs at α = 5 × 10−8, excluded SNPs in strong linkage disequilibrium (LD), and included only SNPs with F-statistics > 10. We used the inverse variance-weighted (IVW) method to estimate causal effects, presenting results as p-values, odds ratios (ORs), and 95% confidence intervals (CIs). Heterogeneity was assessed using Cochrane's Q test and I2 statistics, with I2 < 25% indicating no heterogeneity and I2 < 50% indicating mild heterogeneity. Potential horizontal pleiotropy was evaluated using MR-Egger regression and MR-PRESSO intercept tests. Sensitivity analyses were performed using leave-one-out methods to confirm the robustness of the results. All analyses were conducted using the “TwoSampleMR” package (Version 0.6.8) in R version 4.4.2.
Consequently, we applied the scikit-learn Python package for establishing random forest model. Briefly, the AD clinical stage and the HIS score were recognized as the response variable, and the inflammatory protein levels were selected as the explanatory variables. All samples enrolled in the combined dataset were randomly split into a training set (70%) and a validation set (30%). Subsequently, the SHapley Additive exPlanations (SHAP) values were utilized to visualize key features affecting AD clinical stage and the vascular cognitive burden, thus analyzing the significance of individual features that influence the prediction of the outcome and exhibiting the impact of each vital feature on the final machine learning model. SHAP analysis was employed to complement conventional regression by capturing non-linear associations and potential interactions. This approach revealed individual-level effect heterogeneity, thereby strengthening the evidentiary robustness of key biomarkers. It should be emphasized that SHAP values indicate the marginal contribution of features to model prediction rather than causal effects, with causal relationships being established primarily through MR analysis.
Ethics approval and consent
ADNI was approved by the institutional review boards of all participating sites, and written informed consent was obtained from all participants. The present study is a secondary analysis of publicly available ADNI data and was conducted in accordance with the ADNI Data Use Agreement and publication policies.
Results
Baseline characteristics of the participants
A total of 330 individuals were finally included in this study, comprising 37 NL individuals, 227 MCI individuals and 66 patients with AD. Participants’ demographic characteristics and plasma inflammatory protein levels at baseline are presented in Table 1. Both MCI and AD patients showed a higher proportion of APOE ε4 gene carriers (p < 0.001), while NL individuals possessed a higher proportion of APOE ε2 gene carriers (p < 0.001). However, there was no significant difference in sex, age, or education levels. In terms of inflammatory proteins levels, cortisol, carcinoembryonic antigen (CEA), TNF-α, IL-3, and complement component 3 (C3) differed significantly (overall p < 0.05) across diagnostic groups.
Demographics and clinical characteristics of the participants.
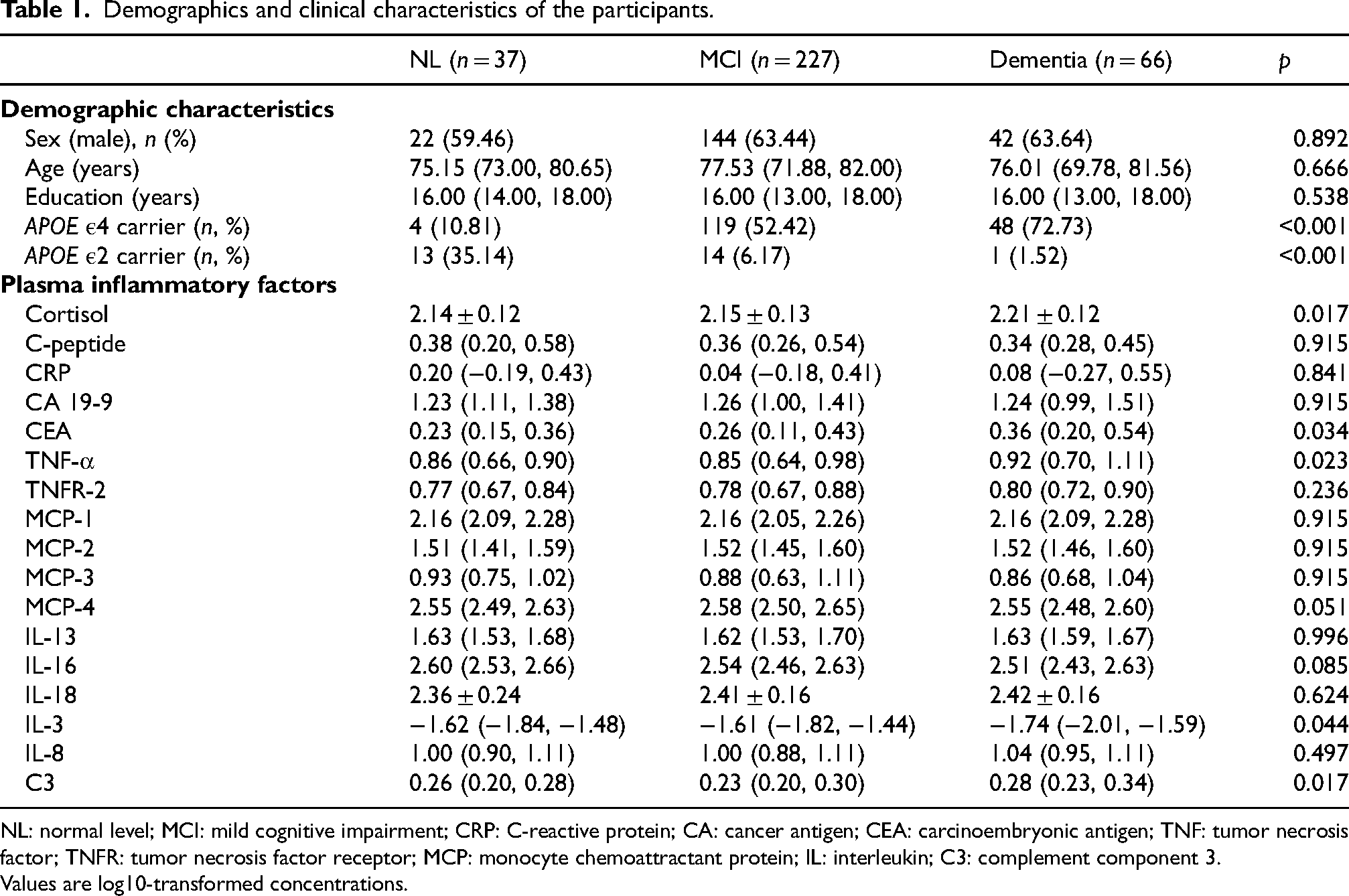
NL: normal level; MCI: mild cognitive impairment; CRP: C-reactive protein; CA: cancer antigen; CEA: carcinoembryonic antigen; TNF: tumor necrosis factor; TNFR: tumor necrosis factor receptor; MCP: monocyte chemoattractant protein; IL: interleukin; C3: complement component 3.
Values are log10-transformed concentrations.
Correlation between inflammatory proteins and the AD clinical stage
To further evaluate the potential relationships between inflammatory proteins and AD, we employed logistic regression models (Table 2). Prior to modeling, plasma protein concentrations were trimmed to remove extreme outliers and standardized to z-scores to facilitate comparability across biomarkers. In univariate logistic regression, we observed a correlation between cortisol (p = 0.001; OR = 1.464; 95% CI: 1.159–1.850), CEA (p = 0.035; OR = 1.290; 95% CI: 1.019–1.634), TNF-α (p = 0.004; OR = 1.421; 95% CI: 1.122–1.799), IL-16 (p = 0.048; OR = 0.790; 95% CI: 0.626–0.998), IL-3 (p = 0.021; OR = 0.762; 95% CI: 0.605–0.961), C3 (p = 0.018; OR = 1.335; 95% CI: 1.051–1.696) and the AD clinical stage after FDR correction. Then we adjusted for covariates including sex, age, education level, and APOE ε2/4 genotype in the multivariate logistic regression analysis. We found that the associations between cortisol (p = 0.015; OR = 1.519; 95% CI: 1.085–2.127), C-peptide (p = 0.026; OR = 0.662; 95% CI: 0.461–0.952), TNFR-2 (p = 0.033; OR = 1.600; 95% CI: 1.038–2.465), IL-16 (p = 0.010; OR = 0.608; 95% CI: 0.417–0.888) and AD clinical stage were significant. Among those four inflammatory proteins related to the AD clinical stage, cortisol and TNF-α were risk factors for AD clinical stage, while C-peptide and IL-16 were protective factors for AD clinical stage.
Correlation between inflammatory proteins and the AD clinical stage.
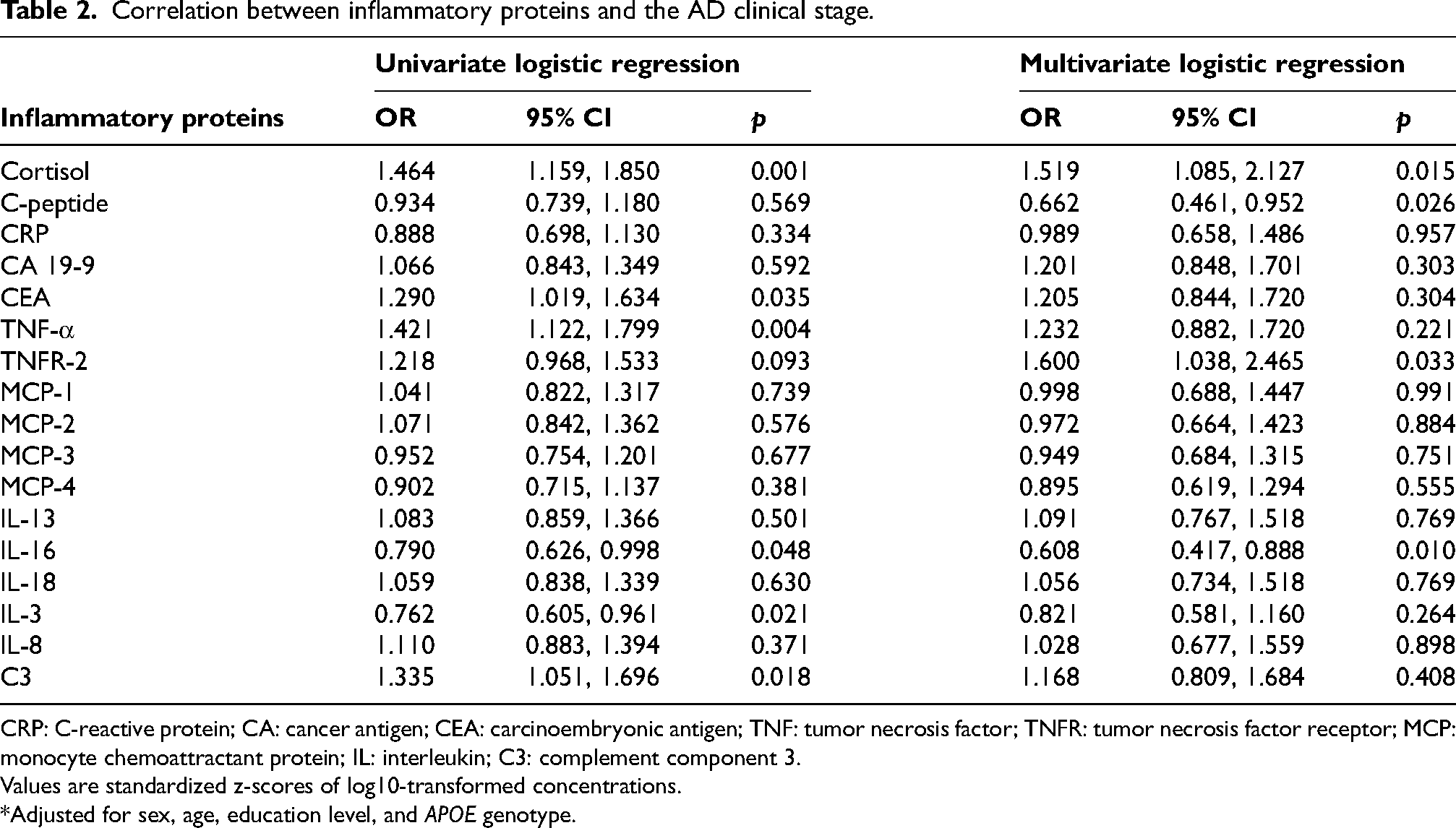
CRP: C-reactive protein; CA: cancer antigen; CEA: carcinoembryonic antigen; TNF: tumor necrosis factor; TNFR: tumor necrosis factor receptor; MCP: monocyte chemoattractant protein; IL: interleukin; C3: complement component 3.
Values are standardized z-scores of log10-transformed concentrations.
*Adjusted for sex, age, education level, and APOE genotype.
Sensitivity analysis using bootstrap resampling
Considering the sample-size imbalances, we performed 2000-case non-parametric bootstrap resampling stratified by diagnosis. Supplemental Table 1 summarizes the mean difference and bias-corrected 95% CI for each pairwise comparison. The sensitivity analysis confirmed that cortisol, TNF-α, TNFR-2, IL-16, IL-3 and C3 exhibited robust and directionally consistent differences across different stages of AD, whereas associations for CRP and IL-18 were not significant, requiring further validation.
Correlation between inflammatory proteins and HIS score
Similarly, we utilized generalized linear models to explore the association between inflammatory proteins and HIS scores which served as a clinical tool to evaluate the vascular cognitive burden in this study (Table 3). Prior to modeling, plasma protein concentrations were trimmed to remove extreme outliers and standardized to z-scores to facilitate comparability across biomarkers. In univariate logistic regression, we observed a correlation between C-peptide (p = 0.003; OR = 1.106; 95% CI: 1.036–1.181), CRP (p = 0.011; OR = 1.095; 95% CI: 1.021–1.174), TNF-α (p = 0.005; OR = 1.103; 95% CI: 1.031–1.180), TNFR-2 (p = 0.001; OR = 1.150; 95% CI: 1.075–1.230), MCP-2 (p = 0.006; OR = 1.105; 95% CI: 1.030–1.185), IL-16 (p = 0.031; OR = 1.080; 95% CI: 1.007–1.157), IL-8 (p = 0.011; OR = 1.093; 95% CI: 1.021–1.169) and the vascular cognitive burden after FDR correction. Then we adjusted for covariates including sex, age, education level, and APOE ε2/4 genotype in the multivariate logistic regression analysis. We found that the associations between C-peptide (p = 0.014; OR = 1.115; 95% CI: 1.095–1.130), CEA (p = 0.034; OR = 1.017; 95% CI: 1.002–1.031), TNFR-2 (p = 0.026; OR = 1.130; 95% CI: 1.016–1.258) and vascular cognitive burden were significant. All of them were risk factors for HIS score, suggesting their strong associations with vascular cognitive burden.
Correlation between inflammatory proteins and HIS score using generalized linear model.
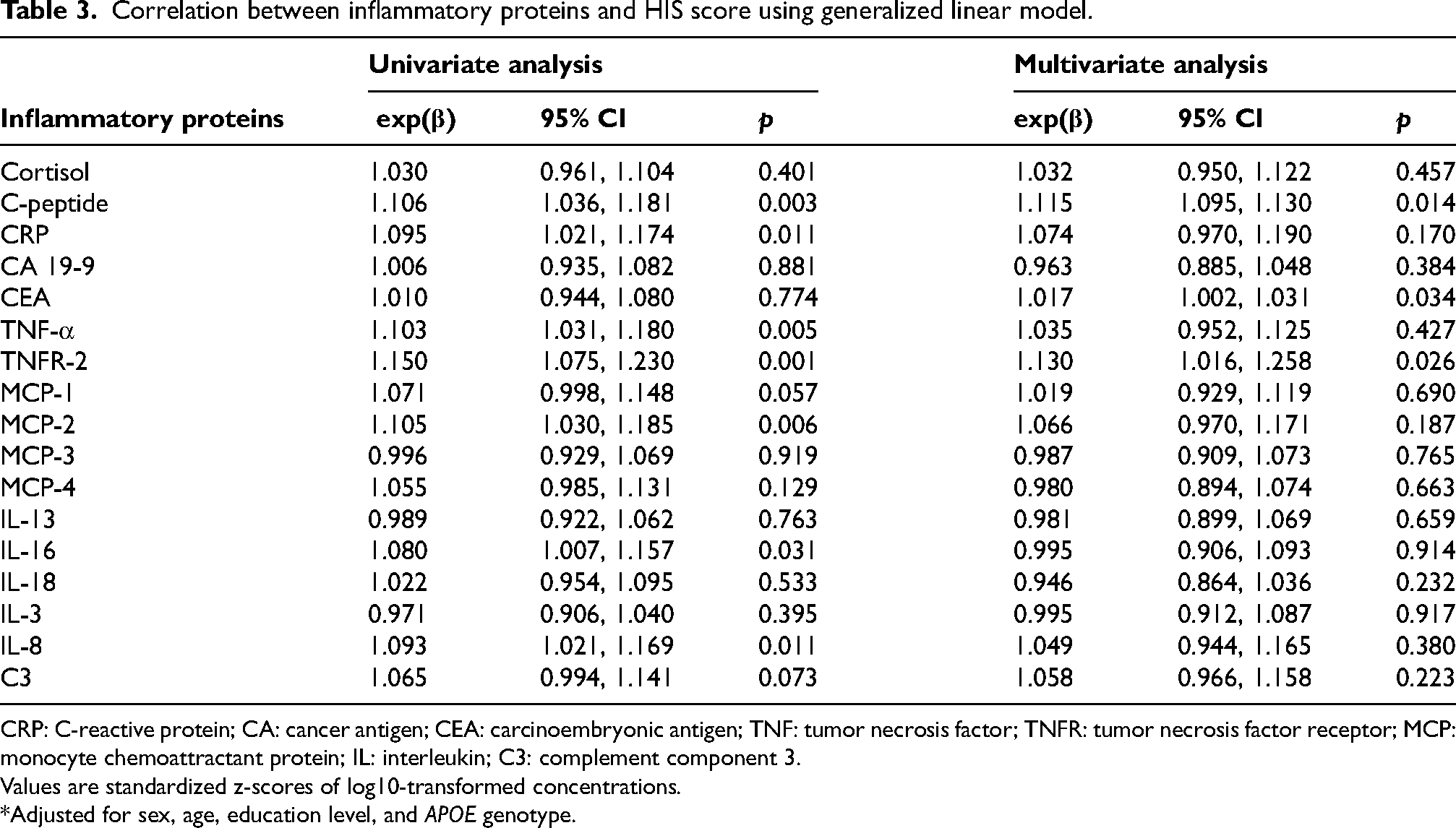
CRP: C-reactive protein; CA: cancer antigen; CEA: carcinoembryonic antigen; TNF: tumor necrosis factor; TNFR: tumor necrosis factor receptor; MCP: monocyte chemoattractant protein; IL: interleukin; C3: complement component 3.
Values are standardized z-scores of log10-transformed concentrations.
*Adjusted for sex, age, education level, and APOE genotype.
The non-linear relationship between inflammatory proteins and dementia
In addition, to further our understanding on the correlation between inflammatory proteins and AD clinical stage, we conducted RCS analyses to assess the non-linear relationships between them (Figure 1). In these analyses, participants were divided into two groups based on their cognitive status: NL individuals and those with MCI or AD. The RCS was applied to the continuous plasma protein levels across their full range, with AD clinical stage as the outcome. When the AD clinical stage was treated as a binary variable, we observed a significant nonlinear relationship only between IL-18 (p for nonlinear = 0.027) and the AD clinical stage among the above eight inflammatory proteins significantly associated with AD clinical stage. The results of other nonsignificant nonlinear relationships are shown in Supplemental Figure 1.
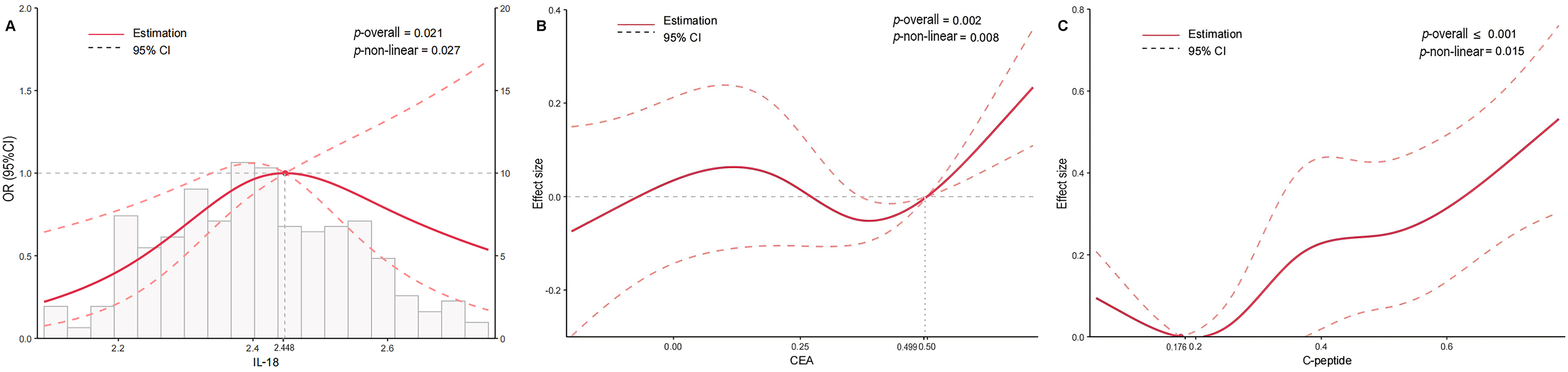
(A) The non-linear relationship between IL-18 and AD. (B) The non-linear relationship between CEA and HIS. (C) The non-linear relationship between C-peptide and HIS. *Adjusted for sex, age, education level, and APOE genotype. AD: Alzheimer's disease; CEA: carcinoembryonic antigen; IL: interleukin.
Likewise, RCS analyses were also performed to evaluate the non-linear relationship between inflammatory proteins and the HIS score (Figure 1). Among the seven inflammatory proteins significantly associated with vascular cognitive burden, only C-peptide (p for nonlinear = 0.015) and CEA (p for nonlinear = 0.008) exhibited significant nonlinear relationships with vascular cognitive burden. The results of other nonsignificant nonlinear relationships are shown in Supplemental Figure 2.
Causal relationship between IL-16 and AD using Mendelian randomization
Subsequently, in order to verify the relationship between inflammatory proteins and AD identified in the ADNI dataset externally, we utilized the GWAS dataset that were independent of the ADNI cohort in combination with MR analysis to further explore the causal relationship between them. Among the above eight inflammatory proteins associated with AD, seven were available for validation analysis in GWAS, but only two genomes, IL-16 and IL-18, identified more than three independent predictors reaching the genome-wide significance threshold (p < 0.001). The analysis results of IL-16 are presented in Figure 2. Of the five MR methods employed, both the weighted median method (p = 0.038; OR = 0.981; 95% CI: 0.964–0.999; Figure 2A) and the IVW method (p = 0.003; OR = 0.981; 95% CI: 0.969–0.994; Figure 2A), which is considered robust against pleiotropy, found a weak negative causal relationship between IL-16 and AD with a strong significance. This finding is consistent with our previous analysis in Table 2, indicating a potential protective effect of the IL-16 against AD. Figure 2B illustrates the scatter plot from the MR analysis, highlighting the relationship between the SNP effect on IL-16 and the SNP effect on AD for each method. The horizontal lines represent the estimated causal effect, and the vertical lines indicate the 95% CIs. The IVW method, represented by the light blue line, suggested a negative causal effect of IL-16 on AD, which is supported by the significant p-value obtained in Figure 2A. The other methods, depicted in different colors, provided less precise estimates, with wider CIs that included the null effect. The forest plot of MR Effect values is shown in Supplemental Figure 3, and the result of sensitivity analysis is shown in Supplemental Figure 4. Sensitivity analyses confirmed the robustness of the primary MR findings, while we observed no significant heterogeneity or evidence of directional pleiotropy. The MR analysis results of IL-18 are shown in Supplemental Figures 5–7. Among the five MR methods employed, none identified a significant causal relationship between IL-18 and AD.
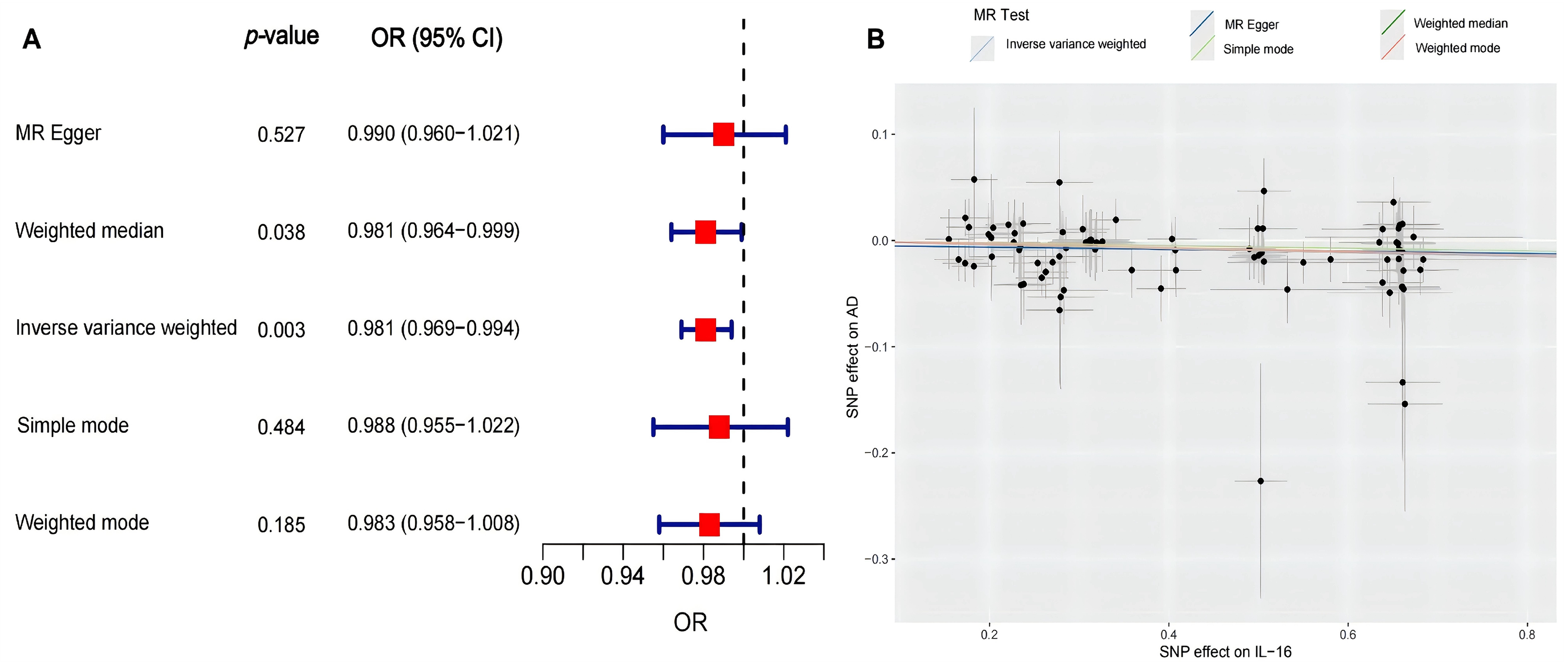
(A) The causal relationship between IL-16 and AD analyzed by different MR methods. (B) The scatter plot of SNP effects on IL-16 and AD. AD: Alzheimer's disease; IL: interleukin; MR: Mendelian randomization; SNP: single nucleotide polymorphism.
To explore the mechanistic complexity underlying the divergent associations of IL-16 with AD, we conducted a competitive bidirectional mediation analysis (Supplemental Figure 8). Results revealed opposing indirect pathways: IL-16 was positively associated with AD via TNFR2 (indirect effect = + 0.33, risk-enhancing), while TNFR2 was negatively associated with AD via IL-16 (indirect effect = –0.35, protective). Notably, IL-16 retained a significant direct protective effect on AD (b = –0.77, p < 0.01) independent of TNFR2, suggesting that the net observational association between IL-16 and AD may reflect the counterbalance of these competing pathways.
SHAP analysis of feature importance
Consequently, we employed the random forest algorithm to build predictive models and utilized SHAP analysis for feature importance and interpretability. The predictive performance metrics of the SHAP-based random forest model are detailed in Supplemental Figure 9. The SHAP plot (Figure 3A) illustrates the impact of different inflammatory proteins on the model output, with the SHAP values indicating the contribution of each feature to predicting the AD clinical stage. The color gradient represents the feature values, with red indicating higher values and blue indicating lower values. Mean absolute SHAP value for each feature is presented in Figure 3B. It reveals that IL-16 demonstrated the highest value at 0.084, followed by CEA at 0.068, and C3 at 0.063, indicating that their significant importance in the predictive outcome of the model among all the inflammatory biomarkers. For IL-16 specifically, while the SHAP values were mostly clustered around zero, indicating a generally neutral effect on the prediction of AD clinical stage, there was a slight skew toward negative values, suggesting a potential minor negative influence when present in higher quantities. In Figure 3C and Figure 3D, the SHAP values demonstrate the contribution of each feature to predicting the vascular cognitive burden, with TNFR-2 ranked as the most important predictive factor, followed by C-peptide and TNF-α, emphasizing their substantial impact on predicting the vascular cognitive burden.
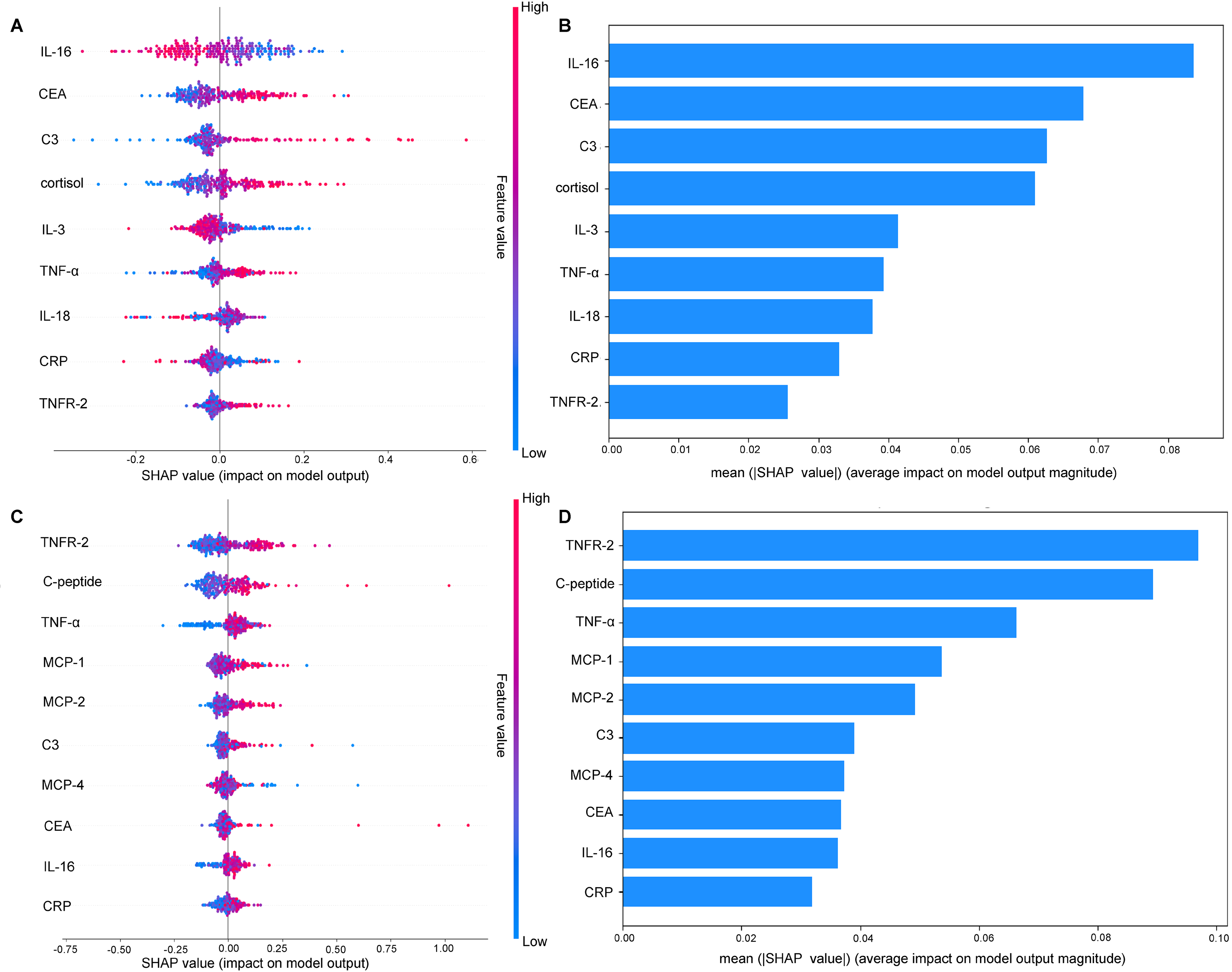
(A) Impact of inflammatory proteins on predictive model for the AD clinical stage. (B) The importance of inflammatory proteins in AD ranked by mean absolute SHAP value. (C) Impact of inflammatory proteins on predictive model for HIS score. (D) The importance of inflammatory proteins in vascular cognitive burden ranked by mean absolute SHAP value. AD: Alzheimer's disease; HIS: Hachinski Ischemia Scale; SHAP: SHapley Additive exPlanations; CRP: C-reactive protein; CA: cancer antigen; CEA: carcinoembryonic antigen; TNF: tumor necrosis factor; TNFR: tumor necrosis factor receptor; MCP: monocyte chemoattractant protein; IL: interleukin; C3: complement component 3.
Discussion
Our main findings were the significant associations between the AD clinical stage and four inflammatory proteins including cortisol, C-peptide, TNFR-2, and IL-16, while the HIS score was significantly associated with three inflammatory proteins including C-peptide, CEA, and TNFR-2. Our findings revealed that cortisol, IL-16 were exclusively associated with AD, while vascular cognitive burden appeared to be exclusively linked to CEA. Our MR results further demonstrated a negative link between AD and IL-16, which later ranked as the most important predictor in the SHAP analysis. Besides, this is the first study to report the nonlinear relationships between IL-18 and AD clinical stage, and between C-peptide, CEA and the vascular cognitive burden based on RCS analyses.
Inflammation has long been implicated in the pathogenesis of both AD and VaD, with evidence suggesting that inflammatory processes play a central role in the development of AD. In AD, cytokines and chemokines can induce neuroinflammation, damage the neuronal environment, and thus contribute to the development of oxidative stress and cell apoptosis, leading to symptoms that are indicative of AD. 10 As for VaD, risk factors for vascular cognitive impairment also could cause inflammation and vascular oxidative stress, which in turn impair the factors that regulate the cerebral circulation, thereby contributing to the vascular cognitive burden. 5 However, the precise mechanisms remain controversial.
In this study, we found that cortisol, C-peptide, TNFR-2, and IL-16 were associated with AD. Despite their distinct roles in the immune and inflammatory responses, they may share several common pathways, which may prolong inflammatory cell survival and contributes to neuroinflammation, thereby accelerating the progression of AD.27–30 In contrast, vascular cognitive burden appeared to be linked to proteins associated with vascular inflammation, including C-peptide, CEA and TNFR-2. These proteins are known to play roles in endothelial dysfunction, atherosclerosis, and cerebral small vessel disease, all of which are critical factors in the development of cardio-cerebrovascular diseases.31–33 The distinct inflammatory profiles observed in AD and VaD suggest that while both conditions involve inflammatory processes, the specific pathways and mechanisms may differ. Additionally, two inflammatory proteins, including C-peptide and TNFR-2, were correlated with both AD and vascular cognitive burden, which is broadly consistent with previous studies, implying that AD and vascular cognitive burden may share some overlapping pathways through certain inflammatory factors.34,35 These findings furnish an exploratory framework for dissecting inflammation-related pathways across the Alzheimer spectrum and the vascular-contribution continuum, offering an insight for future mechanistic and interventional investigations.
Furthermore, the non-linear relationships observed between those dementia-related inflammatory proteins and dementia are particularly intriguing. The nonlinear association between AD and IL-18 suggests that IL-18 may have a dual role in AD pathogenesis, acting as both a pro-inflammatory mediator at low levels and a potential regulatory factor at higher levels. While evidence indicates that IL-18 has been reported to influence amyloid precursor protein processing and Aβ production in experimental systems, its role as a regulatory factor requires further investigation.36,37 Similarly, significant non-linear relationships were observed between HIS and both C-peptide and CEA, indicating that both excessively low and high levels of these two proteins may exert distinct influences on the vascular cognitive burden. Although a study has identified CEA as a potential plasma biomarker for dementia, and the dual effect of C-peptide on cellular activation and atherosclerosis has been reported, this is the first study to propose nonlinear relationships between these two and vascular cognitive burden.35,38 Such non-linear dynamics underscore the complexity of inflammatory processes in dementia and highlight the potential presence of threshold effects, where optimal levels of certain proteins may be protective, while deviations from these levels may be detrimental. This phenomenon has been observed in the association between 25-hydroxyvitamin D and the risk of dementia. 39
Moreover, our MR and SHAP analyses results suggest that IL-16 could potentially act as a potential protective factor against AD. However, this is in contrast to the majority of studies which have reported that higher levels of IL-16 are associated with an increased risk of AD onset.24,40 The competitive bidirectional mediation analysis offers a plausible explanation: IL-16 appears to exert a direct protective effect on AD, consistent with our MR findings; however, this benefit may be partially offset by a parallel pathway wherein IL-16 is positively correlated with TNFR2, which in turn substantially increases AD risk. This competing interplay between protective and risk-enhancing pathways may account for the modest effect size of IL-16's association in our analyses. Nevertheless, further experimental validation is required to substantiate this hypothesis. Also, future studies with larger sample size and more comprehensive genetic data are needed to further elucidate the relationships between dementia and other inflammatory proteins that were not analyzed in this study.
Our study has several strengths. First, the use of a multi-method approach, including generalized linear models, RCS, MR, and SHAP analysis, provides a comprehensive analysis of the relationships between inflammatory proteins and dementia. Second, this study not only investigated the relationships between inflammatory proteins and both AD clinical stage and vascular cognitive burden, but also compared the differences in inflammatory biomarkers between the two conditions, which could aid in more accurate diagnosis and targeted treatment in the future. Also, there is a scarcity of research on the role of inflammatory biomarkers in vascular cognitive burden compared to AD clinical stage. This study systematically explores the relationships between inflammatory proteins and vascular cognitive burden and compares it with AD, filling an important gap in the literature and providing valuable references for understanding the pathogenesis of VaD. Third, our findings on the relationships between HIS and plasma inflammatory proteins not only link inflammation to the vascular cognitive burden, but also offer a novel perspective for further optimizing HIS score. By incorporating scoring rules for plasma inflammatory proteins specifically related to HIS, it may help to improve the accuracy of HIS in differentiating dementia types.
Despite these strengths, our study has its limitations. ADNI participants are not population-representative, and additional restriction to those with complete proteomics and HIS data may have introduced selection bias; therefore, generalizability to broader clinical populations is limited. Also, given the small size of the cognitively normal subgroup and the multiplicity of biomarkers tested, our findings should be interpreted cautiously. Moreover, HIS relies on clinical history and neurological examination without neuroimaging confirmation of cerebrovascular lesions. It serves primarily as a discriminative tool rather than a validated outcome measure for VaD. Thus, our results should be considered hypothesis-generating regarding vascular contributions to cognitive impairment. External validation in VaD-enriched cohorts is essential. Consequently, although we have adjusted for key demographic factors and APOE genotype, residual confounding by vascular comorbidities, medications, lifestyle factors, and site-level differences cannot be excluded and may partly explain the observed associations.
Future studies should aim to validate our findings in larger and more diverse cohorts to enhance the generalizability. Additionally, exploring the causal relationships between inflammatory proteins and VaD using larger genetic datasets will provide a more comprehensive understanding on the role of inflammation in dementia.
Conclusion
This study identified several inflammatory proteins associated with cognitive status and vascular burden. RCS analyses suggested potential non-linear patterns for selected biomarkers, and two-sample MR provided supportive evidence for a modest protective association of IL-16 with AD. However, these findings should be considered hypothesis-generating and require confirmation in independent, well-characterized cohorts. Future studies integrating harmonized proteomics, longitudinal outcomes, and prespecified analytical plans are warranted to clarify clinical utility.
Supplemental Material
sj-docx-1-alz-10.1177_13872877261443713 - Supplemental material for Association of inflammation-related plasma proteins with vascular cognitive burden and Alzheimer's disease: A multi-method study
Supplemental material, sj-docx-1-alz-10.1177_13872877261443713 for Association of inflammation-related plasma proteins with vascular cognitive burden and Alzheimer's disease: A multi-method study by Zhinan Ye, Bingxin Teng, Shiyue Wang, Jinrong Zhu, Jiaxuan Chen, Bo Zhang, Hejia Cai, Ruting Wei, Bohao Li, Xiaoya Xie, Xicheng Yu, Junjie Li, Suwen Huang, Yiyun Weng, Dehao Yang and in Journal of Alzheimer's Disease
Footnotes
Acknowledgements
Data collection and sharing for the ADNI is funded by the National Institute on Aging, United States (National Institutes of Health Grant U19AG024904). The grantee organization is the Northern California Institute for Research and Education. In the past, ADNI has also received funding from the National Institute of Biomedical Imaging and Bioengineering, United States, the Canadian Institutes of Health Research, Canada, and private sector contributions through the Foundation for the National Institutes of Health (FNIH) including generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation, United States; Araclon Biotech; BioClinica, Inc., United States; Biogen, United States; BristolMyers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company, United States; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare, United Kingdom; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck, Denmark; Merck & Co., Inc., United States; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation, United States; Pfizer Inc.; Piramal Imaging; Servier, France; Takeda Pharmaceutical Company, Japan; and Transition Therapeutics. The Canadian Institutes of Health Research provides funds to support ADNI clinical sites in Canada. Private-sector contributions are facilitated by the Foundation for the NIH (![]() ). The grantee organization is the Northern California Institute for Research and Education, and the study was coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory of Neuro Imaging at the University of Southern California.
). The grantee organization is the Northern California Institute for Research and Education, and the study was coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory of Neuro Imaging at the University of Southern California.
Ethical considerations
The study procedures were approved by the institutional review board at each of the ADNI participating sites. All participants provided written informed consent prior to enrolment in the study. This research was conducted ethically in accordance with the World Medical Association Declaration of Helsinki. No further ethics approval was required because of the public characteristics of the data of GWAS.
Consent to participate
Not applicable
Consent for publication
Not applicable
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (No. 82302081), the Zhejiang Provincial Natural Science Foundation of China (No. LQ24H090003), and the National Innovation and Entrepreneurship Training Program for College Students (No. 202510343022).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data supporting the findings of this study are publicly available in ADNI at https://adni.loni.usc.edu. These data were accessed under a license permission granted by the database provider, and their use complies with the terms specified in the original license. Genetic association data for AD are available in GWAS catalog at ![]() .
.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.