
Abstract
Salmonella is known to cause intestinal infections in humans, which can result in symptoms such as diarrhea, vomiting, and, in severe cases, sepsis and death. Isolates of Salmonella from specific populations and regions exhibit varying patterns of antibiotic resistance and genetic characteristics. This study aims to evaluate the diversity of 32 strains of Salmonella isolated from fecal samples of diarrhea patients in Chifeng City, China, between 2021 and 2023, through antibiotic resistance and genomic analysis. Microbroth dilution and whole genome sequencing were employed to investigate antibiotic resistance, multilocus sequence typing, phylogenetic relationship, virulence genes, antibiotic resistance genes, and mobile genetic elements. The antibiotic resistance tests showed 93.75% of Salmonella isolates were resistant to ampicillin, followed by streptomycin (STR) (87.50%) and tetracycline (TET) (56.25%). A total of 32 Salmonella strains were classified into 6 ST types. Virulence gene profiles revealed a relatively high prevalence of the type III secretion system gene cluster. The adhesion-related genes fim, Bcf, and Agf/Csg were prominently represented across all isolates. The antibiotic resistance gene profile showed that the aminoglycoside resistance gene aac(6')-Iaa (100.00%), sulfanilamide resistance gene sul (81.25%), β-lactam resistance gene blaTEM-1B (75.00%), and TET resistance gene tet (56.25%) were more commonly found. Plasmid replicons IncFII(S), IncFIB(S), IncQ1, and IncX1 were frequently identified in these isolates, serving as primary sources of horizontally acquired foreign genes. The most common phage types were Salmon_118970_sal3 (78.13%) and Phage_Gifsy_2 (59.38%). The most frequently observed insertion sequences were IS285 (56.25%) and ISEc39 (56.25%) from the IS256 family. The results indicate a correlation between the resistance phenotype of Salmonella and its genomic characteristics. These findings provide valuable references for the prevention and control of Salmonella, as well as for clinical treatment.
Introduction
Salmonella is a significant pathogenic bacterium responsible for diarrheal diseases (Ramatla et al., 2024). Infections caused by Salmonella typically present as acute headache, fever, vomiting, and diarrhea, which may sometimes be accompanied by blood in the stool, loss of appetite, and general fatigue (Le et al., 2025). In severe cases, these infections can be life-threatening. Salmonella is estimated to cause approximately 9.4 million infections and 115,000 deaths globally each year, with a particularly severe impact in developing countries (Djeghout et al., 2017). Therefore, Salmonella poses substantial threats to human health.
In recent years, the excessive use of antibiotics has resulted in the ongoing emergence and rapid dissemination of novel antibiotic resistance mechanisms, complicating prevention and control efforts. Antibiotic resistance in Salmonella, particularly multidrug resistance (MDR), has emerged as a significant global public health concern. According to relevant surveillance data, the MDR of Salmonella increased from 20% to 30% in the early 1990s and further escalated to 70% in the early 2000s (Mosaddegh et al., 2024). MDR strains, such as S. Typhimurium, S. Enteritidis, and the increasingly prevalent S. Indian, have posed significant public health concerns in recent years (Zelalem et al., 2024; Zhang et al., 2022). Numerous studies have demonstrated a gradual increase in antibiotic resistance among clinical isolates of Salmonella. Both human and non-human sources of resistant strains to ceftriaxone also exhibited resistance to nalidixic acid, sulfamethoxazole (SXT), tetracycline (TET), and ciprofloxacin (CIP) (Kuang et al., 2018). Doma et al. demonstrated that Salmonella isolated from human feces carried plasmids containing resistance genes qnrA, qnrB, and qnrS, which confer resistance to fluoroquinolones (Doma et al., 2020). Among the non-typhoidal Salmonella isolates obtained from the feces of hospitalized patients in Guangzhou, China, 92.16% exhibited resistance to ampicillin (AMP), and 47.06% of these isolates displayed MDR (Liang et al., 2019).
The resistance mechanisms of Salmonella are highly complex and typically arise from the interaction of two or more distinct mechanisms. However, current research predominantly centers on model strains or a limited selection of clinical isolates, clinical application guidelines remain constrained. In this study, we comprehensively analyzed the drug-resistance phenotypes, antibiotic resistance genes (ARGs), virulence genes, and mobile genetic elements (MGE) of Salmonella isolates. By examining the genetic background and antibiotic resistance characteristics, exploring their epidemic characteristics and molecular mechanisms of antibiotic resistance. The findings provide valuable references for the prevention and control of Salmonella, as well as for clinical treatment.
Materials and Methods
Sample source and strain isolation
From March 2022 to November 2023, fecal samples and data were collected from 480 patients diagnosed with unexplained diarrhea who were treated at three sentinel hospitals in Chifeng City. The primary clinical manifestations included defecation occurring three or more times daily, with abnormal stool characteristics such as watery stool, mucus stool, thin stool, or stool containing pus and blood. Fecal samples were collected aseptically, stored in Cary-Blair transport medium (Haibo Biotechnology Co., Ltd, HBPT004) at 4°C, and transported to the laboratory for pathogen isolation and culture within 24 h.
Fresh fecal swabs were inoculated with Selenite Brilliant Green (SBG) sulfanilamide growth solution (Beijing Landbridge Technology Co., LTD., CM244-05) and incubated at 37°C for 18–24 h for pre-cultivation. Subsequently, the bacterial growth solution was transferred onto xylose lysine desoxycholate agar plates (Beijing Landbridge Technology Co., LTD., PB030A) and incubated at 37°C for an additional 18–24 h. More than five suspected Salmonella single colonies were selected and inoculated onto nutrient agar (Beijing Landbridge Technology Co., LTD., PB005A) and triple sugar iron (Beijing Landbridge Technology Co., LTD., M150) for preliminary biochemical reaction identification experiments. Biochemical identification was then conducted using the VITEK Technical Executive Committee (VITEK) 2 Compact automatic microbial identification instrument (Biomeerieux, France), followed by further identification using a Matrix-Assisted Laser Desorption/ Ionization Time of Flight Mass Spectrometry (MALDI-TOF) mass spectrometer (Bruker, microflex LRF).
Identification of serotype
The QIAamp DNA Mini Kit (Qiagen, 51304) was employed to extract nucleic acids from Salmonella strains, and the PCR-probe method was utilized for the preliminary molecular serotype identification of the isolates. The operational procedure followed the instructions provided in the molecular serotype identification kit for Salmonella (Beijing Meizheng Biotechnology Co., LTD., LR706F2), the results were validated using Salmonella diagnostic serum (Ningbo Tianrun Bio-Pharmaceutical Co., LTD., TR101).
Antimicrobial susceptibility
Antimicrobial susceptibility testing was conducted using the minimal inhibitory concentration (MIC) method with the Gram-negative plate (Shanghai Fosun Biological Technology Co., Ltd., A-5). The 16 antimicrobials (11 classes) used were AMP, ampicillin/sulbactam (AMS), TET, meropenem (MEM), polymyxin E (CT), ertapenem (ETP), ceftazidime/avibactam (CZA), tigecycline (TGC), cefotaxime (CTX), ceftazidime (CAZ), CIP, azithromycin (AZI), chloromycetin (CHL), SXT, STR, and amikacin (AMK). For detailed operational procedures, please refer to the literature by Wei et al (2025). The MIC breakpoints of each antimicrobial were used as recommended by the current Clinical and Laboratory Standard Institute (CLSI, 2021) and European Committee on Antimicrobial Susceptibility Testing guidance (ECAST, 2024). Salmonella isolates were defined as MDR when it exhibits resistance to three or more antimicrobial classes (Schwarz et al., 2010). ATCC25922 and ATCC700603 were quality control strains (Nanjing Leche Biotechnology Co., Ltd.).
Whole genome sequencing
Nucleic acid extraction, library construction, and whole genome sequencing
Blood agar medium was utilized for the cultivation of Salmonella, with several typical colonies selected following incubation for 16–18 h at 37°C. The genomic DNA from the samples was extracted using the QIAamp DNA Mini Kit (Qiagen, 51304). The concentration of nucleic acids was quantified using a Qubit 4.0 (Thermo Fisher Scientific), and DNA integrity was assessed via 1% agarose gel electrophoresis. The Ion Xpress™ Plus Fragment Library Kit and Agencourt™ AMPure™ XP Kit (Thermo Fisher Scientific, 4471269) were utilized for genome fragmentation, amplification, purification, and construction of the sequencing library. Preparation of the Ion AmpliSeq template and chip loading was facilitated by the Ion 510™ & Ion 520™ & Ion 530™ Kit-Chef (Thermo Fisher Scientific, A34019), along with the Ion 530™ Chip Kit reagents (Thermo Fisher Scientific, A27764), using the Ion Chef System (Thermo Fisher Scientific). Sequencing was conducted on the Ion GeneStudio S5 (Thermo Fisher Scientific).
Genome assembly and quality control
FastQC was used to assess the quality of read pairs, followed by filtering with the fastX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Reads were filtered using PEAR (https://doi.org/10.1093/bioinformatics/btt593). The merged reads were assembled with SPAdes (https://doi.org/10.1089/cmb.2012.0021) and aligned using BLAST [https://doi.org/10.1016/S0022-2836(05)80360-2]. QUAST (http://cab.cc.spbu.ru/quast/) was utilized to assess the assembly results. The final sequencing data should achieve a minimum of 100× coverage of the genome length of the tested species, with genome coverage of at least 95%, overall coverage of no less than 100x, and the number of scaffolds limited to fewer than 100.
Phylogenetic analysis based on SNP
To better capture the evolutionary relationships among closely related Salmonella isolates, the phylogenetic tree was constructed based on single nucleotide polymorphism (SNP) in the genome. S. Typhimurium LT2 (NC_003197.2) served as the reference genome for comparison. Sequence alignment was conducted using MEGA 11, which also facilitated the construction of a phylogenetic tree. SNP analysis was performed using the online tool CSI Phylogeny 1.4 (Habib et al., 2022), inferring a phylogeny based on the concatenated alignment of high-quality SNPs. The pipeline was run with default parameters, select 10× as the minimum depth at SNP positions, 10% as the minimum relative depth at SNP positions, 10 bp as the minimum distance between SNPs, 30 as the minimum SNP quality, 25 as the minimum read mapping quality, and 1.96 as the minimum Z-score. The maximum likelihood algorithm profile in Newick tree format was generated using FastTree (v2.1) (Price et al., 2010). Interactive Tree Of Life was utilized for the displaying, annotating, and managing of the phylogenetic tree (Letunic and Bork, 2021).
Genome bioinformatics analysis and antibiotic resistance mechanism
The sequence type was determined through multilocus sequence typing (MLST) using the PubMLST scheme (https://pubmlst.org/organisms/salmonella-spp). The NCBI database was utilized to compare, retrieve, and download reference sequences. The comparison and analysis of ARGs were conducted using the Antibiotic Resistance Gene Database (http://arpcard.Mcmaster.ca) and the ResFinder database (https://cge.food.dtu.dk/services/ResFinder/). (Bortolaia et al., 2020; Zankari et al., 2012). The comparison parameters were established with a minimum length of 80% and a similarity threshold of 90%. In conjunction with the results from the antibiotic resistance tests, ARGs were identified and analyzed to investigate the role of key ARGs in the antibiotic resistance mechanisms of Salmonella. The virulence factors and inserted sequences were compared using the Virulence Factors Database (VFDB, http://www.mgc.ac.cn/VFs/) and the ISFinder database (https://github.com/thanhleviet/ISfinder-sequences). The characteristics of plasmids and replicons were analyzed using the PLSDB database (plasma database, https://ccb-microbe.cs.uni-saarland.de/plsdb/). Additionally, the identification and visualization of phage regions were conducted using PHASTER (https://phaster.ca/).
Data statistics
Statistical analyses were conducted using SPSS version 25.0. The χ2 test or Fisher’s exact test was employed for group comparisons, with a significance level set at p < 0.05.
Results
Typing of Salmonella
In this study, 32 strains of Salmonella were isolated, resulting in a detection rate of 6.67% (32/480). In the years 2022 and 2023, 23 strains and 9 strains of Salmonella were detected, respectively. The information of the isolates and their hosts is shown in Supplementary Table S1. The detection rates between the 2 years exhibited significant differences, which were statistically significant (χ2 = 6.563, p = 0.010). The typing results showed that there were 14 strains of S. Typhimurium, 10 strains of S. Enteritidis, 5 strains of S. Dublin, and 3 strains of S. Kentucky.
Antibiotic resistance testing of Salmonella
The results of the antibiotic resistance test indicated that the resistance rate of Salmonella to AMP was 93.75% (30/32), followed by STR at 87.50% and TET at 56.25%. Additionally, the resistance to AMS was noted at 28.13%. It was noted that 3.13% exhibited resistance to the first-line clinical drug CIP. Additionally, 56.25% of the strains were found to be in a mediating state. Furthermore, 15.63% and 6.25% of Salmonella strains demonstrated significant resistance to CTX and CAZ, respectively, both of which are classified as cephalosporins. It was concerning that 23SAL665 isolates were concurrently resistant to both ciprofloxacin and cephalosporins. All strains exhibited sensitivity to MEM, ETP, CZA, TGC, and AMK, with a sensitivity rate of 100.0% (Table 1).
Antimicrobial Resistant Rate of Salmonella Isolates against 16 Antimicrobials (%)

The statistical analysis revealed that 84.38% of Salmonella strains examined in this study were multidrug resistant, with predominant resistance observed against AMP, STR, TET, AMS, and other antibiotics. These strains exhibited resistance to between three and nine different antibiotics, with the highest proportion (17 strains, 53.12%) being resistant to three antibiotics. Additionally, three strains (9.38%) were resistant to four and five antibiotics, respectively, while two strains (6.25%) showed resistance to seven antibiotics. Furthermore, one strain (3.13%) demonstrated resistance to six and eight antibiotics, respectively (Supplementary Fig. S1).
Genome sequencing analysis of Salmonella
The whole genome sequencing of Salmonella isolates has been successfully completed. Based on the results of the QUAST analysis, the genome coverage ranges from 95.06% to 99.166%. The total length of the sequences varied between 4,732,036 and 4,955,105 base pairs, while the GC content ranged from 51.87% to 52.18%. Additionally, the maximum fragment length varied from 409,124 to 1,318,009 base pairs. The GenBank accession and other detailed information regarding the genomic data can be found in Supplementary Table S2.
MLST typing and phylogenetic relationship construction based on SNP
The results showed that 32 Salmonella strains were classified into 6 types, including ST11 (14/32), ST19 (1/32), ST34 (9/32), ST198 (1/32), ST314 (2/32), and ST2066 (5/32). The phylogenetic tree of SNPs was further constructed based on the results of MLST classification. This phylogenetic tree was divided into four branches: 9 ST34 in clade 1, 5 ST2066 and 1 ST19 in clade 2, clade 3 comprised 2 ST314, and 1 ST198, 14 ST11 were distributed in clade 4 (Fig. 1A).
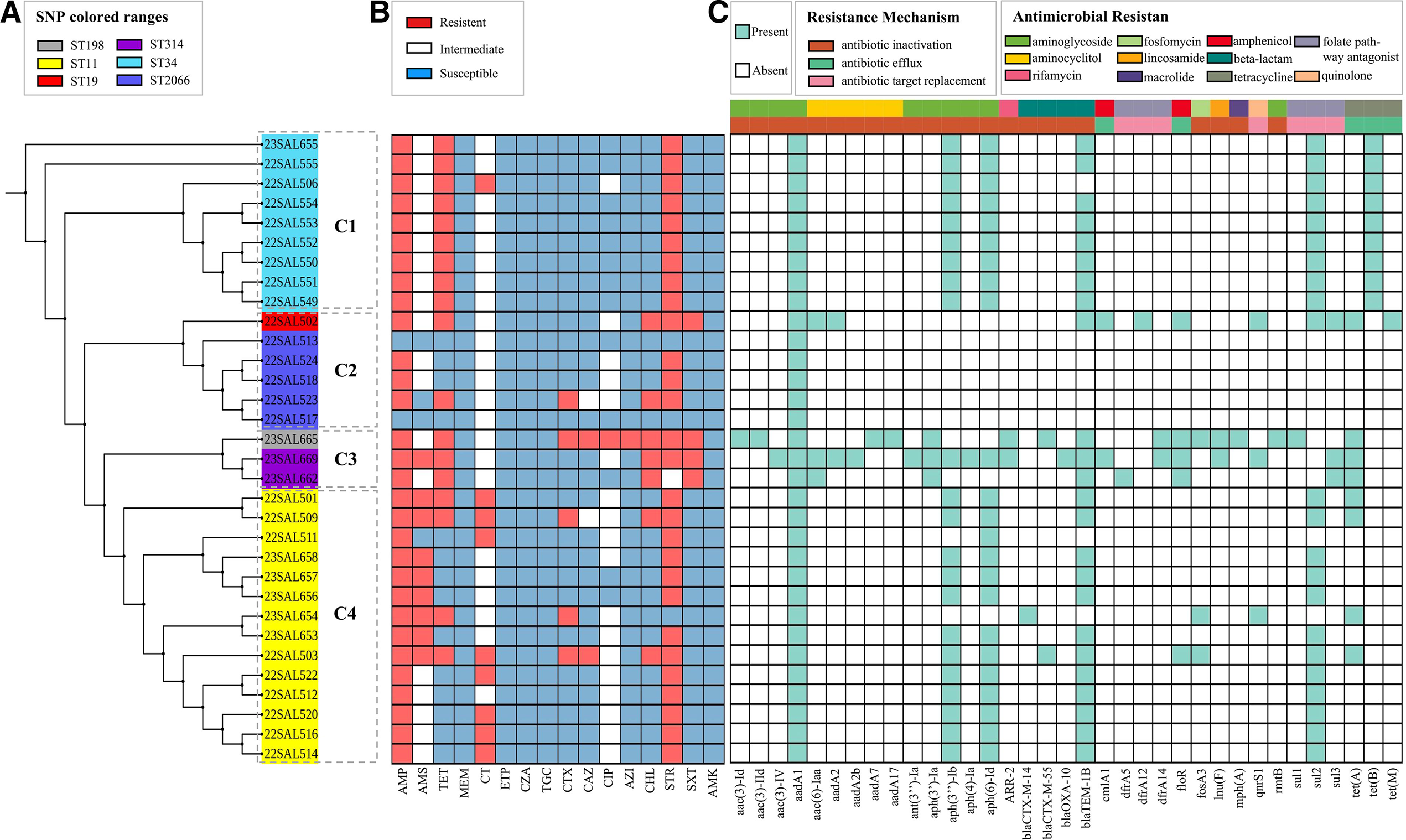
Evolutionary clones of 32 Salmonella isolates.
Analysis of antibiotic resistance pattern and identification of ARG
Whole genome sequencing data from 32 isolates were analyzed using the ResFinder database and compared with the results of antibiotic resistance tests (Fig. 1B). The results indicated that there were 35 ARGs associated with the antibiotic resistance of strains across various drug categories (Fig. 1C). All isolates contained at least one ARG, with the majority related to aminoglycoside resistance. These included aac(3)-Id, aac(3)-IId, aac(3)-IV, aac(6)-Iaa, ant(3'')-Ia, aph(3')-Ia, aph(3′’)-Ib, aph(4)-Ia, aph(6)-Id, and rmtB.
All isolates carried the aminoglycoside acetyltransferase gene aac(6’)-Iaa, which is associated with aminoglycoside resistance. Additionally, 75.00% (n = 24) of the isolates harbored the β-lactam and cephalosporins resistance gene blaTEM-1B, while 71.88% (n = 23) carried the aph(6)-Id gene. Furthermore, 68.75% (n = 22) of the isolates possessed the aminoglycoside resistance gene aph(3'')-Ib. The prevalence of TET resistance genes tet (tet(A), tet(B), tet(M)) was 56.25% (n = 18), and the occurrence of amide alcohol-related genes cmlA1 and floR was 21.88%. The carrying rate of the ciprofloxacin gene qnrS1 was 9.37%. In conjunction with the results of the in vitro antibiotic resistance test (Fig. 1C), the resistance rate of 32 strains to STR, which belongs to the aminoglycosides, was as high as 87.50%. The resistance rate to TET was 56.25%, while the resistance rate to AMP, a penicillin antibiotic, reached 93.75%. The resistance rate to CIP was 3.13%, and the resistance rate to CHL was 21.88%. These results indicate that the expression of various ARGs present in the genome contributes to differing degrees of resistance to their corresponding antibiotics. Notably, genomic data analysis revealed that although the carrying rate of sulfonamide resistance-related genes sul (sul1, sul2, sul3) was as high as 81.25% (n = 26), the resistance rate to the sulfonamide compound SXT observed in the antibiotic resistance test was only 12.50%. For further details on antibiotic resistance, drug categories, and antibiotic resistance mechanisms, please refer to Supplementary Table S3.
Identification of virulence factors
Salmonella exhibits high pathogenicity, making the study of its virulence factors critically important. An analysis of the virulence factors present in the whole genome data of 32 Salmonella isolates revealed a total of 46 factors (see Fig. 2 and Supplementary Table S4). These factors encompass 15 categories, including fimbrial adherence determinants, macrophage-inducible genes, magnesium uptake, non-fimbrial adherence determinants, regulation secretion systems, serum resistance, stress adaptation toxins, adherence, autotransporters, invasion, anaerobic respiration, immune evasion, and antiphagocytosis. Notably, the TTSS (Type III Secretion System) gene clusters related to regulation secretion systems, specifically SPI-1 and SPI-2, contained a high number of genes across all isolates. Additionally, the TTSS-1 translocated effectors gene cluster represented a significant proportion of the total. The fim gene cluster associated with the adhesion system (fimA, fimC, fimD, etc.), the Bcf gene cluster (BcfA, BcfB, BcfC, etc.), and the Agf/Csg gene cluster (CsgA, CsgB, CsgC, etc.) were also highly representative across all isolates.
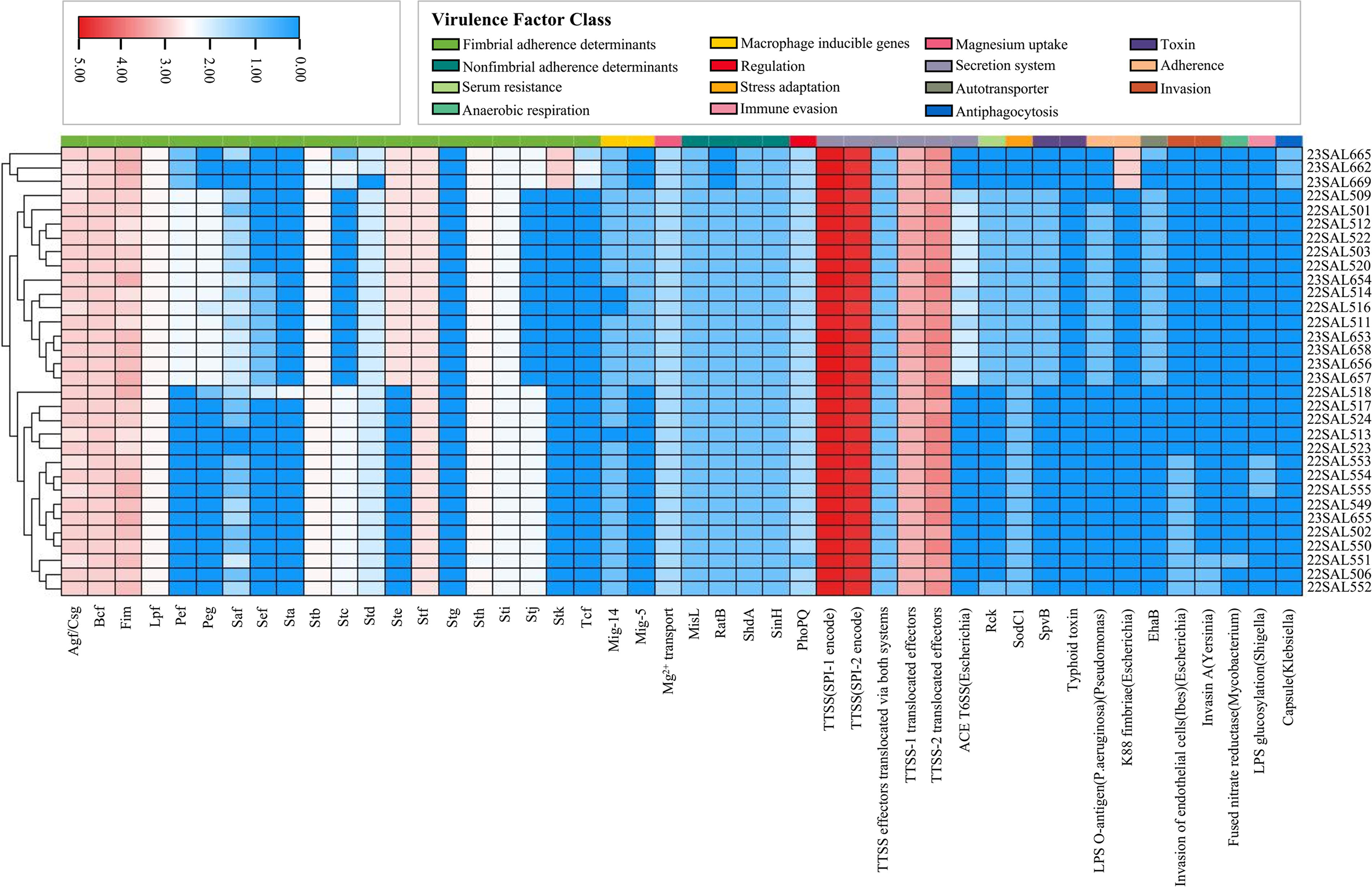
Virulence characteristics of the 32 Salmonella isolates. Different colors at the top of this section represent the class of each virulence factor, including fimbrial adherence determinants (Agf/Csg, Bcf, Fim, Lpf, Pef, Peg, Saf, Sef, Sta, Stb, Stc, Std, Ste, Stf, Stg, Sth, Sti, Stj, Stk, Tcf), nonfimbrial adherence determinants (MisL, RatB, ShdA, SinH), Serum resistance (Rck), Anaerobic respiration (Fused nitrate reductase), Macrophage inducible genes (Mig-14, Mig-5), Regulation (PhoPQ), Stress adaptation (SodC1), Immune evasion (LPS glucosylation), Magnesium uptake (Mg2+ transport), Secretion system (TTSS[SPI-1 encode], TTSS[SPI-2 encode], TTSS effectors translocated via both systems, TTSS-1 translocated effectors, TTSS-2 translocated effectors, ACE T6SS[Escherichia]), autotransporter (EhaB), antiphagocytosis (Capsule[Klebsiella]), toxin (SpvB, Typhoid toxin), Adherence (LPS O-antigen [P.aeruginosa] [Pseudomonas], K88 fimbriae [Escherichia]), invasion (Invasion of endothelial cells[Ibes] [Escherichia], Invasin A [Yersinia]) (as shown in the inset legend). Different colors in the module represent the different copy numbers of each virulence gene (as shown in the inset legend).
Identification of mobile genetic elements
Identification of plasmid replicon
Plasmids serve as the primary carriers of horizontal gene transfer (HGT) in bacteria. In this study, the PLSDB database was utilized to analyze the plasmid replicates found in Salmonella isolates. It was observed that only one isolate (23SAL662) lacked any plasmids, whereas plasmid replicates were prevalent in the other isolates, which collectively harbored a total of 11 plasmids (see Fig. 3A and Supplementary Table S5). Among these, the four most common types of plasmid replicates were IncFII (S), IncFIB (S), IncQ1, and IncX1, with frequencies of 14, 9, 9, and 5, respectively. Conversely, plasmid replicates with lower frequencies included IncX3 (n = 3), Col(pHAD28) (n = 2), ColRNAI (n = 2), Col156 (n = 1), IncFII(pHN7A8) (n = 1), IncHI2 (n = 1), and IncHI2A (n = 1).
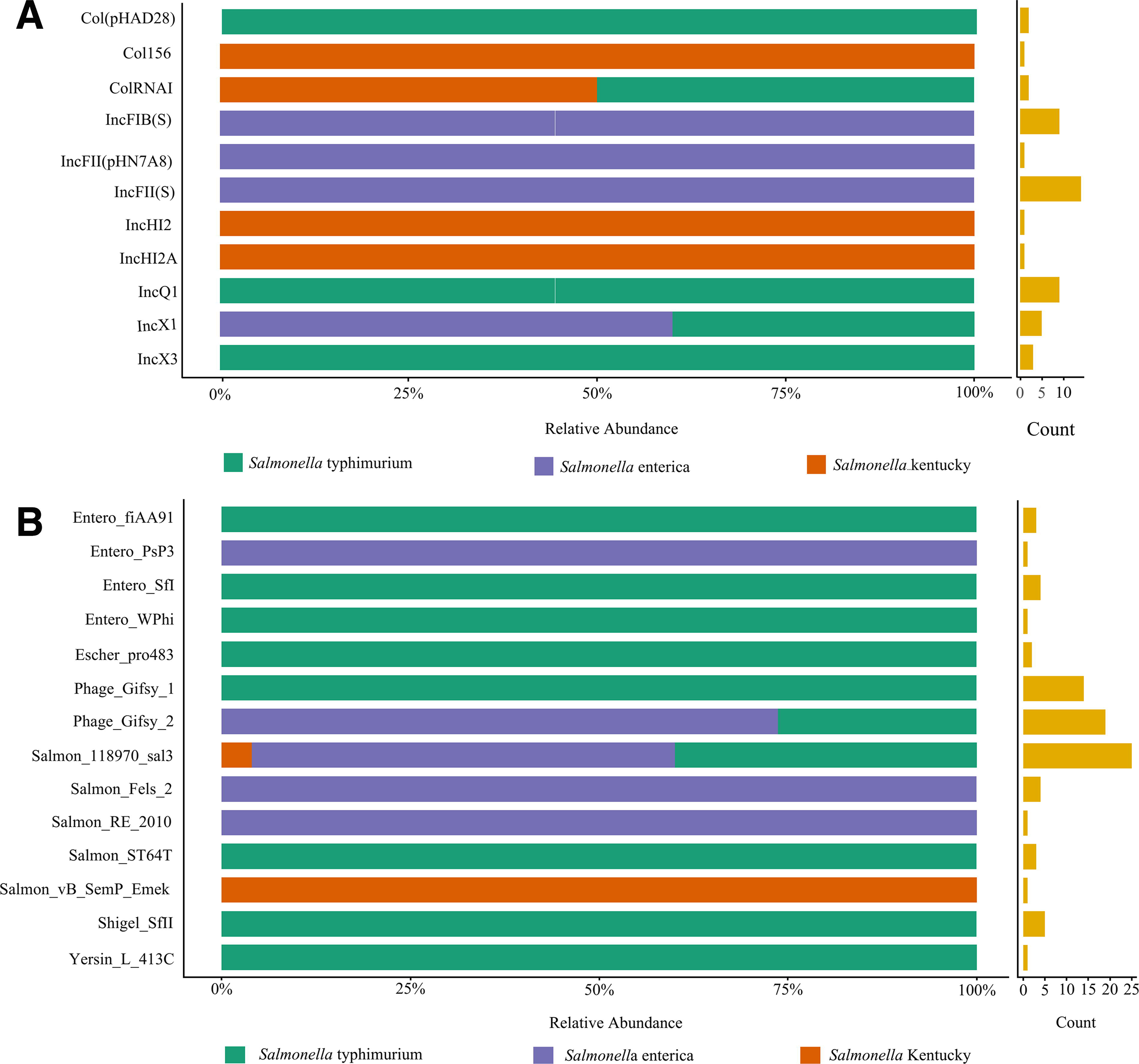
Identification of phage region
The HGT of ARGs mediated by species-specific phages can lead to the emergence of antibiotic-resistant strains, posing a significant threat to public health. In this study, the PHASTER tool was utilized to identify the prophage regions in the isolates. The results indicated that the prophage was prevalent among the isolates. Only one isolate, 23SAL669, contained no complete phage in its genome, as it only harbored two incomplete phages. In contrast, the other isolates exhibited 1–5 complete phage regions, with more than half (17/32, 53.13%) containing three complete phages. All isolates contained 14 distinct types of intact prophages, with the most common being Salmon_118970_sal3 (n = 25, 78.13%), followed by Phage_Gifsy_2 (n = 19, 59.38%) and Phage_Gifsy_1 (n = 14, 43.75%). Additionally, the complete Shigel_SfII phage was detected in five isolates, while Entero_SfI and Salmon_Fels_2 were found in four isolates each. Entero_fiAA91, Salmon_ST64T, and Escher_pro483 were detected in three and two isolates, respectively. Furthermore, Entero_WPhi, Entero_PsP3, Salmon_RE_2010, Salmon_vB_SemP_Emek, and Yersin_L_413C were identified in only one sample. (Fig. 3B and Supplementary Table S6).
Identification of insertion sequence
The characteristics of mobile genetic element insertion sequences in 32 Salmonella isolates were analyzed using ISfinder. A total of 44 insertion sequences were identified (Fig. 4, Supplementary Table S7), comprising 14 members of the IS1 family, 10 members of the IS3 family, and 4 members of the IS4 family. The IS6 family included six members, while the IS200/605 and Tn3 families each contained three members. The IS256 and IS110 families each consisted of two members. The insertion sequences with the highest frequency were IS285 (n = 18) and ISEc39 (n = 18) from the IS256 family, followed by IS1230B (n = 14) from the IS3 family and ISSty2 (n = 14) from the Tn3 family. Notably, the 22SAL506 isolate exhibited the largest number of insertion sequences, totaling 25 distinct types, with the members of the IS1 family being the most abundant.
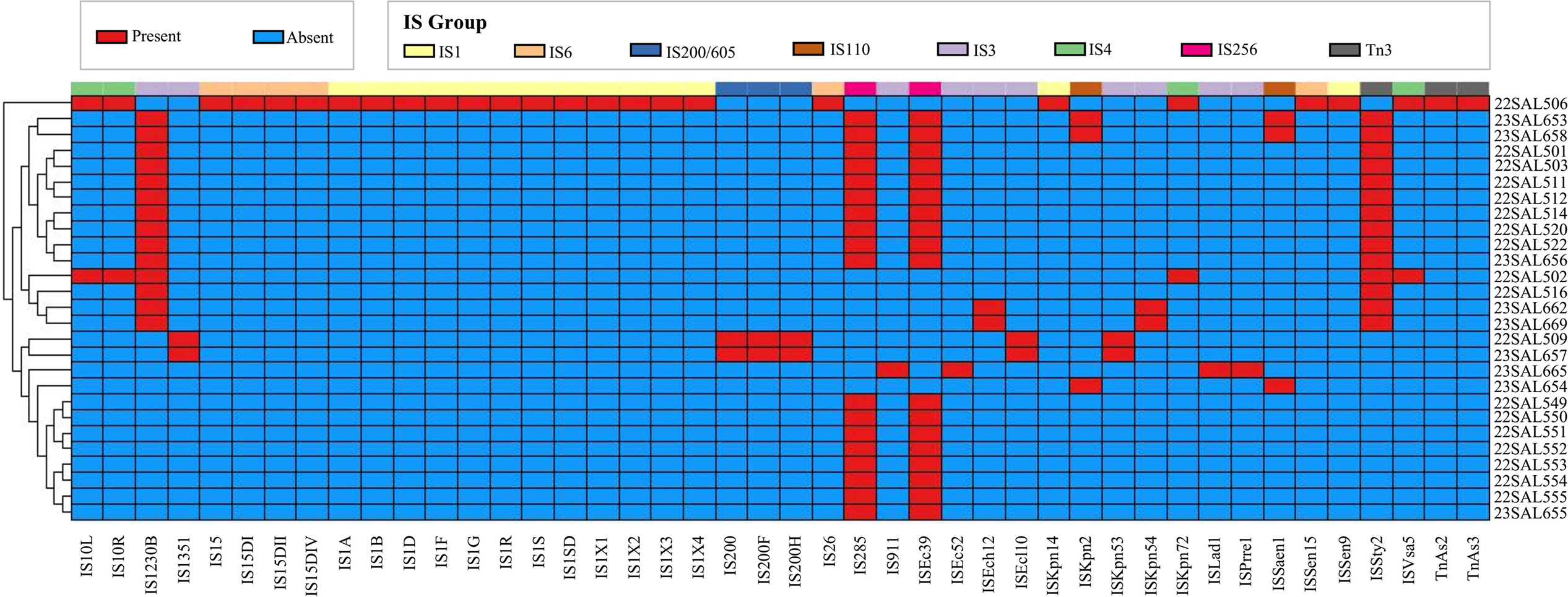
Insertion sequence characteristics of the 32 Salmonella isolates. Different colors at the top of this section represent the group of each IS member, including IS1 (IS1A, IS1B, IS1D, IS1F, IS1G, IS1R, IS1S, IS1SD, IS1X1, IS1X2, IS1X3, IS1X4, ISKpn14, ISSen9), IS6 (IS15, IS15DI, IS15DII, IS15DIV, IS26, ISSen15), IS200/605 (IS200, IS200F, IS200H), IS110 (ISKpn2, ISSaen1), IS3 (IS1230B, IS1351, IS911, ISEc52, ISEch12, ISEcl10, ISKpn53, ISKpn54, ISLad1, ISPrre1), IS4 (IS10L, IS10R, ISKpn72, ISVsa5), IS256 (IS285, ISEc39), Tn3 (ISSty2, TnAs2, TnAs3) (as shown in the inset legend). Different colors in the module represent the presence or absence of each IS (as shown in the inset legend).
Discussion
This study provides a comprehensive genomic and phenotypic characterization of Salmonella isolates from diarrheal patients in Chifeng City, revealing a high prevalence of MDR facilitated by a diverse array of MGEs. Our findings underscore a significant clinical challenge and offer insights into the molecular mechanisms driving antibiotic resistance in this region.
The detection rate of Salmonella (6.67%) in Chifeng aligns with, though is slightly lower than, rates reported in other Chinese regions such as Shanghai (8.3%) (Qi et al., 2016), establishing a baseline for the local epidemiology. The predominance of S. Typhimurium is consistent with Liang et al. (2015) data, highlighting the serotypes of greatest clinical concern. Critically, 84.38% of the isolates were MDR, with disconcerting resistance rates to AMP, STR, and TET. The identification of resistance to critically important cephalosporins (CTX and CAZ) and fluoroquinolones (CIP), including one isolate (23SAL665) co-resistant to both, has direct and grave implications for clinical practice in China. This co-resistance pattern severely limits therapeutic options, potentially rendering frontline treatments like ciprofloxacin and third-generation cephalosporins ineffective for severe infections, thereby necessitating the use of last-resort carbapenems, to which all our isolates remained susceptible.
Genomic analysis confirmed a correlation between the observed resistance phenotypes and the carriage of corresponding ARGs. The high prevalence of aminoglycoside resistance genes (e.g., aac(6')-Iaa, aph(6)-Id, aph(3'')-Ib) corresponded with high STR resistance. Similarly, the presence of blaTEM-1B (75% of isolates) underpinned the widespread ampicillin resistance. However, a critical finding was the discrepancy between the high carriage rate of sulfonamide resistance genes (sul1, sul2, sul3; 81.25%) and the relatively low phenotypic resistance to trimethoprim-SXT (12.50%). This suggests that while the genetic potential for resistance is widespread, these genes may be poorly expressed or regulated in the absence of selective pressure, a nuance that would be missed by relying solely on genomic predictions.
A critical analysis of the cephalosporin resistance gene environment reveals the potential role of IS in resistance dissemination. The high prevalence of blaTEM-1B warrants attention, and MGE analysis identified a rich repertoire of IS elements, including members of the IS1, IS3, and Tn3 families, which are frequently associated with the mobilization of ARGs (Partridge et al., 2018). For instance, the IS256 family (identified here as IS285 and ISEc39) is known to create strong promoters that can upregulate adjacent genes and has been linked to MDR and biofilm formation in other pathogens (Hennig and Ziebuhr, 2010). The co-localization of such IS elements upstream or downstream of blaTEM-1B on plasmids or in the chromosome could facilitate its excision, circularization, and subsequent transposition to other MGEs, such as the prevalent IncFII(S) and IncX1 plasmids we identified (Hansen et al., 2019). Future studies employing complete genome sequencing will be essential to precisely map the genetic environment of key resistance genes like blaTEM-1B and confirm the involvement of specific IS elements in their mobilization.
The potential for HGT is further underscored by the diversity of MGEs we detected. The high-frequency plasmid replicons IncFII(S), IncFIB(S), and IncX1 have been previously implicated in the global spread of ARGs, including qnrS1, mcr, and various β-lactamase genes (Johnson and Nolan, 2009). The presence of the qnrS1 gene in 9.37% of our isolates, located on such mobilizable plasmids, is a key molecular explanation for the reduced susceptibility to ciprofloxacin observed in 56.25% of strains. Furthermore, the ubiquitous prophages, particularly Salmon_118970_sal3 and the virulence-associated Gifsy-1 and Gifsy-2, represent another vector for genetic exchange. Phage transduction has been demonstrated to transfer ARGs like blaCTX-M and tet(M) (Colomer-Lluch et al., 2014), and Gifsy-2 is a known carrier of virulence factors such as sodCI (Golubeva and Slauch, 2006). The interplay between these MGEs creates a highly dynamic genome, accelerating the acquisition and dissemination of both resistance and virulence determinants.
In a broader comparative context, our findings from Chifeng contribute to the alarming national and global trend of increasing antibiotic resistance in Salmonella. The resistance profiles we observed, particularly to cephalosporins and fluoroquinolones, are consistent with reports from other parts of China and Asia (Fu et al., 2020; Sriyapai et al., 2021). This convergence of data highlights a systemic challenge that requires coordinated national antibiotic stewardship and surveillance programs. For clinical practice in China, our data argue for a shift in empirical treatment guidelines for severe diarrheal syndromes in regions with similar resistance patterns. While fluoroquinolones and extended-spectrum cephalosporins remain empirical mainstays, their local efficacy cannot be assumed. The high in vitro susceptibility to AZI (not tested here but a common alternative) and carbapenems should be considered for targeted therapy, emphasizing the critical role of routine antimicrobial susceptibility testing to guide treatment decisions and preserve the efficacy of last-resort agents.
Conclusions
This study is the first to provide genomics and phenotypic description data of Salmonella strains in patients with diarrhea in Chifeng City. A high prevalence of MDR strains (84.38%), exhibiting alarming resistance rates to AMP (93.75%), STR (87.50%), and TET (56.25%). Resistance to key frontline clinical agents, including cephalosporins and ciprofloxacin, was observed, with one isolate showing co-resistance limiting therapeutic options severely. Genomic analysis showed that most phenotypes confirmed in this study correlated with their ARGs, such as blaTEM-1B and aminoglycoside resistance genes. The high carriage of sulfonamide resistance genes (sul), however, and low phenotypic resistance, signifies a gap between genetics and phenotypic traits. The results of the study reveal that different MGEs, including commonly occurring plasmid replicons (IncFII(S), IncFIB(S), IncX1), insertion sequences (like IS256 family), and prophages, play an important role in the horizontal transfer of ARGs and virulence factors. Through further analysis of the isolates, they were found to have important virulence factors such as TTSS-1 and TTSS-2, which makes them high risk. The research from Chifeng shows that Salmonella are increasingly developing antibiotic resistance. It is vital to carry out antibiotic susceptibility testing on a routine basis to reinforce monitoring and coordination of antibiotic management to the serious threat of drug resistance.
Authors’ Contributions
C.W.: Conceptualization, data curation, project administration, methodology, writing—original draft. F.Y.: Project administration, methodology, and writing—original draft. Y.B.: Investigation, protocol development, and writing—review and editing. H.Y.: Investigation, writing—review and editing. L.L.: Conceptualization and funding acquisition. H.L.: Protocol development and writing—review and editing. P.P.: Investigation and validation. Y.Z.: Software, writing—review and editing. Q.C.: Writing—review and editing. All authors read and approved the final draft of the article.
Footnotes
Author Disclosure Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding Information
This study was funded by Natural Science Foundation Project of Inner Mongolia (2025QN08109), Chifeng Yulong Talent Project (2024).
Ethics Statement
The studies involving human participants were reviewed by the Chifeng Center for Disease Control and Prevention and were in compliance with relevant laws and regulations. Approval has been granted by the Ethics Review Committee (Ethical Application Ref: 2024-zr-006). The patients/participants provided written informed consent to participate in this study, and key patient information has been anonymized and protected.
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.