
Abstract
Vibrio vulnificus, a major human-pathogenic Vibrio species, causes foodborne or wound-related infections with a globally rising incidence. The study aimed to characterize the molecular characteristics of V. vulnificus to support the prevention, control, and clinical treatment of its infection. A total of 63 V. vulnificus isolates from seafood and patients in Wenzhou were subjected to sequencing, bioinformatic analysis, and antimicrobial susceptibility testing. Single nucleotide polymorphism and multilocus sequence typing analysis of 100 isolates (63 from this study, 37 from the NCBI database) were performed to assess genetic characteristics. Nine key virulence genes, associated with adhesion, RTX toxin, and T6SS, and eight antimicrobial resistant genes (ARGs) were identified. All isolates were resistant to colistin (CT), with 74.6%, 58.7%, and 3.2% resistance to streptomycin, amikacin, and nalidixic acid, respectively, and were susceptible to 12 other tested antimicrobials. V. vulnificus infection cases in this study showed a significant demographic skew toward males aged ≥50 years. The isolates exhibited genetic diversity while sharing virulence genes and ARGs. In silico detected ARGs could not accurately predict in vitro antimicrobial resistance phenotypes, so extreme caution is required when using ARGs detection results to guide clinical anti-infective medication. This study provides basic genomic and phenotypic data of local V. vulnificus isolates, which may serve as a reference for local surveillance and clinical empirical treatment of V. vulnificus infection.
Keywords
Introduction
Vibrio vulnificus (V. vulnificus) is a lethal Gram-negative pathogenic bacterium that primarily causes wound and gastrointestinal infections (Walter, 2023). Wound infection manifests as local redness, swelling, and pain, which can progress to severe myositis and tissue necrosis, while gastrointestinal infection typically presents with diarrhea, nausea, vomiting, abdominal pain, fever, and chills (Muzembo et al., 2024; Walter, 2023). This pathogen is naturally distributed in warm coastal and estuarine waters, especially high-salinity environments (Walter, 2023). Human infections mainly occur via consumption of contaminated seafood or wound exposure to contaminated water/seafood products, with individuals with underlying diseases and children at elevated infection risk (Muzembo et al., 2024).
V. vulnificus is conventionally divided into three biotypes based on biochemical characteristics (Naknaen et al., 2024). Biotype 1 is the primary causative agent of human infections, biotype 2 mainly infects eels, and biotype 3 exhibits combined phenotypic features of biotypes 1 and 2 (Naknaen et al., 2024). For genetic subtyping, vcg typing is widely used to distinguish clinical and environmental strains, with most clinical isolates carrying the vcgC genotype (Ma et al., 2024; Naknaen et al., 2024). Multilocus sequence typing (MLST) and multiple-locus variable-number tandem repeat analysis further enable classification of V. vulnificus into distinct clonal groups and sequence types, revealing its extensive genetic diversity (Naknaen et al., 2024; Prithvisagar et al., 2023).
V. vulnificus carries a wide repertoire of virulence genes, including vvhA, rtxA, plpA, cps, hutA, vctC, vscC2, and VP_RS21620, which act synergistically to drive its pathogenesis during infection (Choi and Choi, 2022; Ma et al., 2024; Prithvisagar et al., 2023; Wang et al., 2023). These virulence factors are mainly involved in exotoxin production, immune evasion, iron acquisition, and effector delivery (Choi and Choi, 2022). Specifically, the vvhA-encoded hemolysin induces host cell death and inflammatory responses, while the rtxA-encoded MARTX toxin exerts strong cytotoxic and cytopathic effects (Choi and Choi, 2022; Ma et al., 2024). The phospholipase plpA triggers epithelial cell necrosis and erythrocyte lysis, acting synergistically with MARTX toxin to exacerbate tissue damage (Choi and Choi, 2022). Capsular polysaccharide synthesis genes (cps) confer resistance to serum bactericidal activity and host phagocytosis (Wang et al., 2023), while iron acquisition-related genes (hutA and vctC) enable V. vulnificus to scavenge host iron for survival and proliferation (Prithvisagar et al., 2023). In addition, type III secretion system 2 (T3SS2)-associated genes (vscC2 and VP_RS21620) encode an effector delivery apparatus that directly injects virulence effectors into host cells to enhance pathogenicity (Ma et al., 2024).
Antimicrobial resistance (AMR) profiles of V. vulnificus exhibit significant geographical heterogeneity. A study in China reported that 66.7% of V. vulnificus isolates were multidrug-resistant (Xu et al., 2024), whereas studies in the United States showed low and stable AMR rates, with most isolates susceptible to ciprofloxacin (Morgado et al., 2024). In India, seafood-derived isolates harbored AMR genes but remained susceptible to common antibiotics, including amikacin (AMK), azithromycin (AZI), and cefotaxime in susceptibility assays (Prithvisagar et al., 2023), while a Korean study found that most isolates were CT-resistant and all carried cyclic AMP receptor protein (CRP)-related resistance genes (Lee et al., 2024). These regional discrepancies highlight the critical importance of local AMR surveillance for V. vulnificus.
WGS has emerged as a powerful, widely used tool for public health surveillance of bacterial pathogens, enabling comprehensive characterization of the epidemic status, transmission dynamics, and evolutionary selection of virulence and AMR genes in V. vulnificus. In this study, we performed WGS and systematic genomic analysis of V. vulnificus isolates recovered from seafood and patients in Wenzhou, China. We comprehensively analyzed the genetic polymorphism, virulence gene profiles, and AMR characteristics of these isolates to provide a scientific basis for in-depth understanding and precise prevention and control of V. vulnificus infections in this region.
Materials and Methods
Ethical approval
This study complied with the Declaration of Helsinki and China’s Measures for the Ethical Review of Biomedical Research Involving Human Subjects. The study protocol, including the application for waiver of informed consent for retrospective clinical isolate data and residual specimens, was approved by the Medical Ethics Committee of Wenzhou Center for Disease Control and Prevention (Ethics ID: EC-20250107-1022; Approval No. LLSC-2025-004).
Strain collection, isolation, and identification
A total of 63 non-duplicate V. vulnificus isolates were collected in Wenzhou from 2012 to 2023. Of these, 13 clinical isolates were obtained from 5 local sentinel hospitals undertaking statutory infectious disease surveillance, and 50 foodborne isolates were recovered from routine active foodborne pathogen surveillance. The surveillance program collected 200–600 aquatic product samples annually, including bivalve mollusks, marine fish, conch, and soft-shelled turtles sourced from farmers’ markets and retail supermarkets. All isolates were preserved at −80°C with beads.
All experimental procedures strictly followed the National Food Safety Standard of the People’s Republic of China (GB 4789.44). Modified cellobiose-polymyxin B-CT agar (Hopebio, Qingdao, China) was used for selective isolation. Species identification was confirmed via the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS, Bruker Daltonics, Bremen, Germany).
Whole genome sequencing and bioinformatics analysis
Total genomic DNA was extracted using a Bacterial Genomic DNA Extraction Kit (Tiangen, Beijing, China), with concentration, purity, and integrity verified via a NanoDrop One spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and agarose gel electrophoresis. WGS was performed on the Illumina MiSeq platform (Illumina Inc., San Diego, CA, USA) with a paired-end 150 bp strategy and a target sequencing depth of 100×. Raw reads were quality-filtered using FastQC v0.12.1, Fastp v0.24.0, QUAST v5.3.0, and CheckM v1.2.2 (Supplementary Table S1), and de novo genome assembly was conducted via SPAdes v4.0.0. All raw sequencing data have been deposited in the National Microbiology Data Center (NMDC, https://nmdc.cn/) under project number NMDC10019786.
A total of 100 V. vulnificus isolates were analyzed, including 63 isolates from this study and 37 NCBI-derived genomes (inclusion criteria: complete metadata, CheckM completeness ≥98%, and contamination ≤1%). The V. vulnificus type strain ATCC 27562 (GCF_002224265.1) was used as the reference for phylogenetic analysis. Single nucleotide polymorphism (SNP) detection and filtering were performed with Snippy v4.6.0. Recombination regions were removed using Gubbins v3.4.3, followed by neighbor-joining tree construction with FastTree v2.2.0, and the final tree was visualized via iTOL (https://itol.embl.de/). MLST was conducted using mlst v2.32.2 with the curated V. vulnificus scheme from PubMLST (https://pubmlst.org/). AMR and virulence factor genes were identified using Abricate v1.0.1 against the CARD, ResFinder, and VFDB databases, with a stringent threshold of ≥80% identity and ≥80% coverage.
Antimicrobial susceptibility testing
Antimicrobial susceptibility testing (AST) was performed via the broth microdilution method using A-5 type aerobic Gram-negative bacilli AST panels (Fosun Diagnostics Technology Co., Ltd., Changsha, China), in strict accordance with Clinical and Laboratory Standards Institute (CLSI) M45-A3 and M100 34th Edition guidelines. The panels covered 17 clinically and epidemiologically relevant antimicrobial agents for V. vulnificus, including β-lactams, tetracyclines, quinolones, aminoglycosides, polypeptides, phenicols, macrolides, and sulfonamides. The minimum inhibitory concentration of each agent was interpreted using Vibrio-specific breakpoints from CLSI M45-A3, with non-enteric Gram-negative criteria from CLSI M100 34th Edition applied for agents without species-specific thresholds. ATCC 25922, ATCC 27853, ATCC29213, ATCC29212, and ATCC 700603 were included as quality control strains.
Statistical analysis
Statistical analysis was performed using SPSS v26.0 (IBM Corp., Armonk, NY, USA). All categorical variables (gender, age, source, and onset season) were reported as frequencies and percentages with 95% exact Clopper-Pearson confidence intervals. A two-sided exact binomial test was used for binary variables to test deviations from equal distribution, and an exact multinomial goodness-of-fit test was applied for multinomial variables given the small sample size. Statistical significance was set at a two-tailed p < 0.05.
Results
Infection status of V. vulnificus in patients
Of the 13 V. vulnificus infected patients (Supplementary Table S2), 11 were male (84.6%, 95% CI: 54.6%–98.1%) and 12 were aged ≥ 50 years (92.3%, 95% CI: 64.0%–99.8%). The exact binomial test showed a significant skewed distribution toward male and older age among this case series (p < 0.05). For the 13 clinical isolates, 6 were from blood, 5 from feces, and 2 from wound samples, with no significant difference in gene profiles across isolation sources (Table 1).
Infection Status of V. vulnificus in Patients
Molecular types and population evolution analysis of V. vulnificus
SNP and MLST were performed on 100 V. vulnificus isolates to assess their genetic characteristics. A phylogenetic tree (Fig. 1) was subsequently constructed based on genome-wide SNP variants, with a tree scale bar of 100,000 representing the number of SNP differences between isolates. All isolates were categorized into three groups according to their isolation source: Homo sapiens, food, and environmental isolates, and no significant source-specific clustering was observed across the phylogenetic tree. Geographically, the isolates covered nine countries and regions across Asia, Europe, and North America, and the majority of novel sequence types were identified in Wenzhou local isolates from this study. MLST results revealed extensive ST diversity among the 100 isolates, including both globally reported known STs and multiple novel STs, with no dominant clonal lineage detected. Collectively, these results demonstrate that V. vulnificus isolates in this study exhibit high genetic diversity, with no significant clonal aggregation during its endemic circulation in Wenzhou. These findings supplement the basic population genetic data of V. vulnificus in the coastal areas of eastern China and may provide a reference for the optimization of local V. vulnificus molecular surveillance schemes.
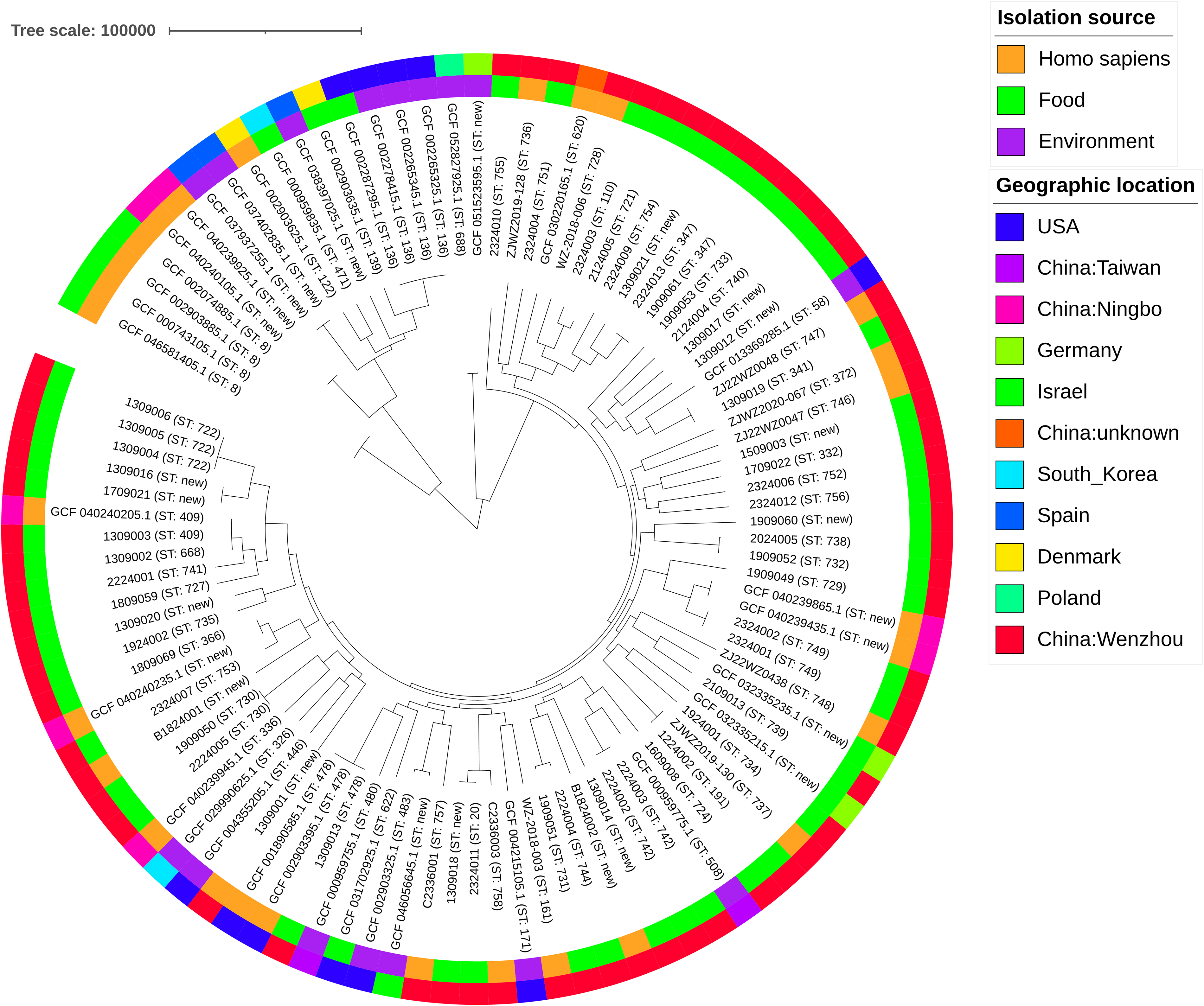
SNP-based phylogenetic tree of 100 V. vulnificus isolates, including 63 isolates from this study and 37 strains from NCBI database.
Molecular characteristics of virulence genes and antimicrobial resistance genes
Genomic alignment of the 63 V. vulnificus isolates against the VFDB identified a total of nine virulence genes, namely IlpA, ompU, rtxB, rtxC, hcp-2, vasD, VCA0109, vipA/mglA, and vipB/mglB (Fig. 2). All isolates were positive for IlpA, ompU, rtxB, and rtxC, while only three isolates harbored the other five genes associated with the type VI secretion system (T6SS). Screening against the ResFinder and CARD identified eight ARGs across the 63 isolates, with the distribution as follows: tet (34) (58/63, 92.1%), sul2 (1/63, 1.6%), CRP (8/63, 12.7%), ermC (1/63, 1.6%), dfrA31 (2/63, 3.2%), dfrA6 (3/63, 4.8%), qnrS5 (1/63, 1.6%), and blaCARB-7 (1/63, 1.6%) (Fig. 3).
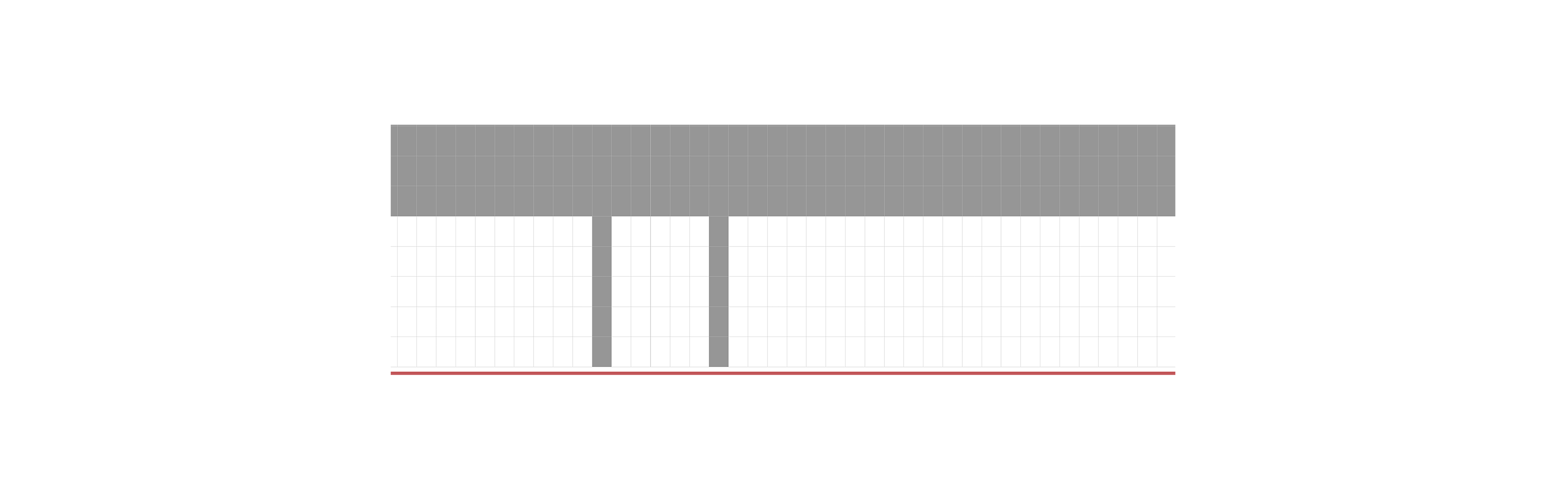
Distribution of putative virulence genes harbored by V. vulnificus isolates in Wenzhou.
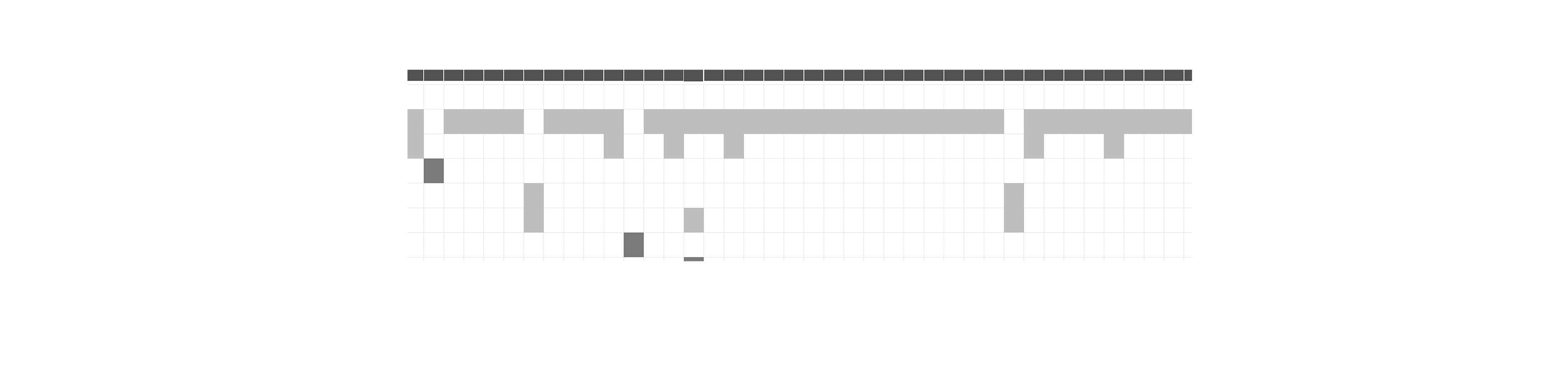
Distribution of putative ARGs harbored by V. vulnificus isolates in Wenzhou.
Antimicrobial resistance genotype-phenotype correlation
The 63 V. vulnificus isolates exhibited 100% resistance to CT, followed by streptomycin (STR, 47/63, 74.6%), amikacin (AMK, 37/63, 58.7%), and nalidixic acid (NAL, 2/63, 3.2%). AZI was not applicable for AST of this species, and all isolates were susceptible to the other 12 tested antimicrobials. (Supplementary Table S3).
Genotype and phenotype correlation analysis revealed a low match rate between ARGs and resistance phenotypes in the 63 V. vulnificus isolates. No matched ARGs were detected in CT-resistant, STR-resistant, AMK-resistant, or NAL-resistant isolates, while the only isolate carrying the quinolone resistance gene qnrS5 was susceptible to NAL. The most prevalent gene tet(34) and all other detected ARGs were identified in isolates with susceptible phenotypes to the corresponding antimicrobial agents.
Discussion
While the prevalence of V. vulnificus infections has remained relatively low, the associated mortality rate is alarmingly high, and identifying high-risk populations is critical for targeted infection prevention and control (Meyer et al., 2022). In this study, we observed a significant skewness in gender and age distribution among 13 V. vulnificus-infected patients in Wenzhou, China, consistent with the well-documented high susceptibility of male and elderly individuals to this infection (Jones and Oliver, 2009). This striking male predominance is mainly attributed to the protective effect of estrogen against V. vulnificus-induced endotoxic shock (Merkel et al., 2001), while the increased infection risk in the elderly is closely related to the higher prevalence of predisposing underlying diseases and impaired innate immune function in this population (Centers for Disease Control and Prevention (CDC), 1993). However, the primary limitation of this study is the small sample size of only 13 cases, which results in insufficient statistical power to validate gender and age susceptibility to V. vulnificus infection, precludes stratified analysis of confounding factors, and limits the external generalizability of our single-center findings. Further validation in large, multicenter epidemiological studies is required.
Seafood, particularly raw or undercooked oysters, is the dominant transmission vehicle for foodborne V. vulnificus infection, with a confirmed causal link to both mild gastrointestinal illness and fatal sepsis (Kling et al., 2022). As a natural inhabitant of warm coastal waters, V. vulnificus causes widespread contamination in seafood (Warner and Oliver, 2008), and clinical case data have directly linked seafood consumption to lethal systemic infection (Xu et al., 2023). These findings are consistent with the consensus that seafood is the main transmission vehicle of foodborne V. vulnificus infection, highlighting the importance of continuous local surveillance of V. vulnificus contamination in seafood for the prevention and control of local foodborne infection.
In our study, SNP analysis and MLST based on housekeeping genes were performed for V. vulnificus isolates, which revealed extensive genetic diversity and significant genomic heterogeneity among the strains, consistent with previous reports of V. vulnificus isolates from Shenzhen and Ningbo, China (Xu et al., 2023, 2024). Phylogenetic analysis based on core SNPs showed that the clinical isolate from Shenzhen was distantly related to sympatric oyster-derived strains but clustered with isolates from Taiwan, China (Xu et al., 2023), while MLST analysis of Ningbo clinical isolates identified distinct evolutionary branches with no complete correlation between phylogenetic clustering and phenotypic characteristics (Xu et al., 2024). Further integrated phylogenetic analysis with global strains showed that neither geographical origin nor isolation source drove the clustering of V. vulnificus. Thus, further studies are needed to elucidate the key factors shaping the distribution of V. vulnificus to better understand its ecological distribution and transmission patterns.
In this study, IlpA, ompU, rtxB, and rtxC were universally detected in all isolates, while five T6SS-related genes were present in only three strains. As previously reported, IlpA encodes an immunogenic lipoprotein driving TLR2-mediated NF-κB activation and robust proinflammatory cytokine production, the mechanism of V. vulnificus-induced septic shock and high mortality (Goo et al., 2007), while ompU encodes a fibronectin-binding outer membrane protein mediating bacterial adhesion to host cells and extracellular matrix, a prerequisite for successful colonization and invasive infection (Goo et al., 2006). The ubiquitous presence of rtxB and rtxC further underscores the essential role of the RTX toxin system in V. vulnificus virulence, as rtxC encodes the toxin maturation activator, rtxB encodes the ABC transporter for toxin secretion, which together regulate delivery of the key effector MARTX toxin mediating host cell damage and tissue necrosis (Lin et al., 1999). In contrast to these virulence genes, the low prevalence of T6SS-related genes indicates T6SS is not an essential determinant of basal pathogenicity in local isolates, and previous studies have linked intact T6SS to enhanced serum survival and anti-complement activity of V. vulnificus, suggesting this system may confer additional virulence advantages, including immune evasion and interbacterial competition for the few T6SS-positive strains and contribute to differential virulence phenotypes among isolate (Zhang et al., 2021). Collectively, the pathogenesis of local isolates may be a synergistic process mediated by these highly conserved virulence factors. ompU mediates initial host adhesion, the rtxB/rtxC-dependent RTX system regulates host tissue invasion related toxin secretion, and IlpA is associated with the induction of host inflammatory responses. The universal presence of these four genes in all isolates suggests that they may be potential candidate markers for the molecular detection of local V. vulnificus isolates, and their functional roles in pathogenicity need to be verified by further in vitro and in vivo experiments.
The study systematically analyzed the correlation between ARG profiles and in vitro AST phenotypes of 63 V. vulnificus isolates from Wenzhou, to characterize the AMR molecular features of local strains and provide laboratory reference for local clinical empirical anti-infective treatment. All isolates showed 100% resistance to CT, with 74.6%, 58.7% and 3.2% resistance to STR, AMK and NAL, respectively, and were susceptible to the other 12 tested antimicrobials. The aligns with reports that most global V. vulnificus strains remain susceptible to first-line clinical antibiotics (Carmona-Salido et al., 2026; Morgado et al., 2024; Sharshar and Azab, 2008; Tang et al., 2010), while the high aminoglycoside resistance rate exhibits regional specificity, similar to aquatic isolates from diseased prawns (Sharshar and Azab, 2008). Eight ARGs were identified, with tet(34) being the most prevalent, and other ARGs had carriage rates below 5%, consistent with the widespread distribution of tet and blaCARB family genes in global V. vulnificus populations (Carmona-Salido et al., 2026). Notably, there was nearly no concordance between in silico acquired ARGs and in vitro AST phenotypes of V. vulnificus isolates. Universal CT resistance with no matched acquired ARGs was the most prominent discrepancy, consistent with previous global studies (Carmona-Salido et al., 2026; Morgado et al., 2024; Zanetti et al., 2001). We cautiously infer that the observed resistance may be driven by chromosomal mutations outside our detection scope, while ARGs in susceptible isolates may be non-functional or silent. These findings confirm that in silico ARGs detection alone cannot reliably predict resistance phenotypes, and standardized phenotypic AST remains essential for clinical treatment. In addition, limitations of our analysis include restricted ARGs screening scope, lack of ARG functional validation, and the inability of in vitro AST to reflect in vivo efficacy, which warrant further functional genomic studies.
Conclusions
In conclusion, the distribution of V. vulnificus infection cases in this study showed a statistically significant skew towards males aged ≥50 years. The isolates exhibited high genetic heterogeneity, yet shared virulence genes and ARGs despite sporadic local infections. Nine key virulence genes related to bacterial adhesion, RTX toxin-mediated host cell damage, and T6SS-associated functions, as well as eight ARGs were identified, among which tet(34) was the most prevalent. Notably, in silico detected ARGs could not accurately predict the in vitro AMR phenotypes of local isolates. Thus, extreme caution is required when using ARGs detection results to guide clinical anti-infective medication, and standardized phenotypic AST remains the essential basis for optimizing clinical treatment regimens for V. vulnificus infection. These findings provide basic data and new insights into the genomic characteristics, virulence and AMR profiles of local V. vulnificus isolates in Wenzhou, highlighting the importance of continuous local surveillance to track the genetic evolution and resistance changes of V. vulnificus strains.
Authors’ Contributions
X.Z.: Methodology (equal); project administration (lead); visualization (equal); writing-original draft (equal); writing—review and editing (lead). G.Z.: Software (lead); visualization (equal); writing—original draft (equal). Y.L.: Conceptualization (lead); investigation(lead); visualization(equal); software (supporting); writing—review and editing (supporting). A.X.: Data curation (lead); formal analysis (lead); resources (supporting); writing—original draft (equal). H.L.: Methodology (equal); writing—original draft (equal). R.Z.: Methodology (equal); writing—original draft (equal). H.L.: Funding acquisition (lead); conceptualization (supporting); methodology (equal); supervision (lead); validation (lead); writing—review and editing (supporting).
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.