
Abstract
Amiodarone, a common and effective antiarrhythmic drug, has been reported to have anti-inflammatory effects such as reducing the activation and movement of neutrophils. However, its effects on human T cells remain unclear. The aim of this study was to elucidate the effects and possible underlying mechanisms of amiodarone on human T cells. We isolated human primary T cells from the peripheral blood of healthy volunteers and performed enzyme-linked immunosorbent assay (ELISA), flow cytometry, electrophoretic mobility shift assay, luciferase assay, and Western blotting to evaluate the modulatory effects of amiodarone on human T cells. We found that amiodarone dose dependently inhibited the production of cytokines, including interleukin-2 (IL-2), IL-4, tumor necrosis factor-alpha, and interferon-gamma in activated human T cells. By flow cytometry, we demonstrated that amiodarone suppressed the expression of IL-2 receptor-alpha (CD25) and CD69, the cell surface markers of activated T cells. Moreover, molecular investigations revealed that amiodarone down-regulated activator protein-1 (AP-1) and nuclear factor kappa-B (NF-κB) DNA-binding activities in activated human T cells and also inhibited DNA binding and transcriptional activities of both AP-1 and NF-κB in Jurkat cells. Finally, by Western blotting, we showed that amiodarone reduced the activation of c-Jun NH2-terminal protein kinase and P38 mitogen-activated protein kinase, and suppressed stimuli-induced I-kappa B-alpha degradation in activated human T cells. Through regulation of AP-1 and NF-κB signaling, amiodarone inhibits cytokine production and T cell activation. These results show the pleiotropic effects of amiodarone on human T cells and suggest its therapeutic potential in inflammation-related cardiovascular disorders.
Introduction
Atrial fibrillation (Af) is one of the most common arrhythmias in clinical practice, accounting for approximately one-third of admissions resulting from cardiac rhythm disturbances, and about 1% of the adult population in the United States has been reported to suffer from Af.1–3 Af frequently coexists with heart failure, affecting about 30% of all individuals with heart failure.4–7 Moreover, Af is a common clinical complication of acute coronary syndrome. In the Cooperative Cardiovascular Project, Af was found to be present in 22% of 106,780 persons aged 65 years and older with acute myocardial infarction. 8 Additionally, in the Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin Therapy trial, Af was found to be present in 6.4% of 9432 patients with non-ST segment elevation myocardial infarction or unstable angina. 9 It has also been reported that Af increases the risk of mortality after myocardial infarction or coronary artery bypass grafting.10,11 These results highlight the importance of Af and its close association with heart failure and acute coronary syndrome.
Although the mechanisms and specific factors leading to this kind of atrial arrhythmia have not been completely determined, previous studies have reported on signaling transduction systems, inflammation, oxidative stress, atrial stretching, and/or ischemia as factors related to the cascade of events causing Af.3,12–15 Bruins et al. first indicated the relationship of Af with an inflammatory process, after patients who developed Af after coronary artery bypass surgery were found to have increased levels of plasma C-reactive protein. 16 More recently, increasing evidence suggests that inflammation is a risk factor for developing new-onset Af. 17
Inflammation plays an important role in the pathogenesis of various diseases, and its characteristics include redness, heat, pain, and swelling, which result from local responses of immune, vascular, and parenchymal cells to infection or injury. 18 Interestingly, during the inflammatory cascade, T cells are known to be a key factor in the progression of atherosclerosis, and they have been linked to acute onset of coronary events. 19 Activated T cells, especially CD4+ T-helper type 1 cells, are major producers of interferon-γ (IFN-γ), which is known to induce the expression of class II major histocompatibility complex antigens on macrophages and vascular smooth muscle cells and to promote atherosclerosis. 20 It is therefore reasonable to hypothesize that the activation of T cells plays a crucial role in the pathogenesis of both Af and atherosclerosis.
Amiodarone is the most effective antiarrhythmic drug for preventing the recurrence of Af and has limited proarrhythmic toxicity.7,21 It is categorized as a class III antiarrhythmia agent (a multichannel blocker) and also has the ability to block alpha- and beta-receptors in cardiac cells. 22 Clinically, it is used to treat various kinds of ventricular and atrial arrhythmias, including suppression of ventricular tachycardia and complex arrhythmias associated with heart disease.23–25 Intriguingly, many pleiotropic effects of amiodarone have been reported, with the most important being to reduce inflammation. 22 Wilson et al. suggested that amiodarone inhibits the phospholipase C signaling pathway which plays a major role in the production of inflammatory mediators in arachidonic acid metabolism. 26 Moreover, amiodarone has been reported to modify phospholipid metabolism in Jurkat cells and to have phospholipidogenic effects on CD3+ T lymphocytes.27,28 Furthermore, amiodarone has been reported to reduce the production of tumor necrosis factor-alpha (TNF-α) in lipopolysaccharide-stimulated human mononuclear cells. 29 All of these studies point to the anti-inflammatory nature of amiodarone in treating cardiovascular disorders.
In this study, we investigated whether one of the effects of amiodarone on slowing and even reversing the incidence of Af in ischemic heart disease is by reducing activated T cell-related inflammation. Our novel findings demonstrate that amiodarone effectively reduces T cell activation and suppresses the production of inflammatory cytokines in human primary T cells. The mechanisms are likely to be mediated through activator protein-1 (AP-1) and nuclear factor kappa-B (NF-κB) signaling pathways. These results suggest that amiodarone has a therapeutic potential in treating inflammation-related cardiovascular disorders.
Materials and methods
Preparation of amiodarone
The purified form of amiodarone (provided by Sanofi-Synthelabo Pharmaceutical) was dissolved in ethanol. The defined concentrations were added to culture media with immune cells to test its effects. The final concentrations in the media were about 1–5 µM.
Isolation of human primary T cells from human blood
Human peripheral blood mononuclear cells were collected from whole blood buffy coat of healthy volunteers as described previously (more than 40 men or women participants with an age from 30 to 60 years). 30 The layer of purified mononuclear cells was collected and incubated with antibodies including L243 (anti-DR; American Type Culture Collection (ATCC), Rockville, MD), OKMI (anti-CD11b; ATCC), and LM2 (anti-MacI; ATCC) for 30 min at 4℃. The cells were then washed with medium containing 0.1% fetal bovine serum and incubated with magnetic beads conjugated with goat anti-mouse IgG (R&D, Minneapolis, MN). The antibody-stained cells were then removed with a magnet. Following a repeat of the above procedures, T cells were obtained with a purity of more than 98% as determined by the percentage of CD3+ cells in flow cytometry (Beckton Dickinson, Mountain View, CA).
Cell treatment and stimulation
For cell activation, the following stimuli were used: 5–20 ng/mL of phorbol 12-myristate 13-acetate (PMA, Sigma, St. Louis, MO), 1 µM of ionomycin (Sigma), 10 µg/mL of immobilized anti-CD3 mAb (OKT3, ATCC), l µg/mL of soluble anti-CD-28 mAb (clone 9.3, kindly provided by Di-Carl June, Naval Institute, Bethesda, NIH), and 2 ng/mL of TNF-α (Sigma, USA).
Measurement of nonspecific cytotoxicity of the drugs
Several assays were used to examine drug cytotoxicity on the cells. The release of lactate dehydrogenase (LDH), as an indicator of damage to the plasma membrane and cell death, was measured according to the assay manufacturer’s instructions (Roche, Indianapolis, IN, USA). The percent of cytotoxicity was calculated as ([sample value – medium control]/[high control – medium control]) × 100, where the sample values were the averages of the absorbance values from triplicate measurements of the indicated dose of amiodarone or vehicle-treated cell culture supernatants after the subtraction of the absorbance values from the background control. The average absorbance values of untreated cell culture supernatants, used as the medium control, were calculated in a similar manner. An equal number of cells treated with 1% Triton X-100 was used as the high control. 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assays were performed as previously described.31,32 In brief, T cells at 1 × 106 mL−1 in 100 µL volume were incubated in the presence or absence of amiodarone for 24 h. Twenty-five microliters of MTT (5 mg/mL in H2O) was then added, and the cells were incubated at 37℃ for 2 h followed by the addition of 100 µL of lysis buffer containing 20% sodium dodecyl sulfate and 50% dimethylformamide. After incubation at 37℃ for another 6 h, the amount of dissolved reduced MTT crystals was measured with an enzyme-linked immunosorbent assay (ELISA) reader (Dynatech, Chantilly, VA).
Cytokine production assay
The determination of cytokine concentrations was performed according to our previous report. 30 Briefly, a 96-well flat-bottom plate was coated with anti-cytokine mAb (100 µL at 4 mg/mL) in phosphate-buffered saline (PBS) pH 7.3 at room temperature overnight. The plate was then washed with PBS containing 0.05% Tween 20 (PBS-T) three times, followed by incubation with a blocking solution containing 1% bovine serum albumin, 5% sucrose, and 0.05% NaN3 in PBS for more than 1 h. After washing with PBS-T, the collected 100 µL of supernatant was added into each well for 24 h. The plates were then washed with PBS-T three times and then incubated with biotinylated anticytokine detection antibodies (l00 µL at 12.5 ng/mL) for 2 h at room temperature. Following washing, 100 µL of streptavidin horseradish peroxidase (1:2000 dilution of a 1.25 mg/mL solution) was added and incubated for 20 min at room temperature. After washing three times, 100 µL of substrate solution containing a 1:1 mixture of H2O2 and tetramethylbenzidine was added and incubated for another 20 min at room temperature. The reaction was stopped by adding stop solution, and the cytokine concentrations were measured with a microplate reader (Dynatech).
Measurement of cell surface molecule expressions
Human peripheral blood T cells at a concentration of 1 × 106 mL−1 were preincubated with various concentrations of amiodarone for 2 h. After adding stimuli for another 24 h, the cells were collected and washed with PBS. After washing, the cells were stained with fluorescein isothiocyanate (FITC)-conjugated anti-interleukin-2 (IL-2) receptor alpha (anti-IL-2Rα, anti-CD25) and anti-CD69 monoclonal antibodies (mAbs) or FITC-conjugated isotype-matched mAb (PharMingen, San Diego, LA), and the expressions of these surface molecules were determined with a flow cytometer (Becton Dickinson).
Nuclear extract preparation
Nuclear extracts were prepared according to our previous study. 33 Briefly, the treated cells were left at 4℃ in 70 µL of buffer A (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.9, 10 mM KCl, 1.5 mM MgC12, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), and 3.3 µg/mL aprotinin) for 15 min with occasional gentle vortexing. The swollen cells were then centrifuged at 15,000 r/min for 3 min. After removal of the supernatants (cytoplasmic extract), the pelleted nuclei were washed with 70 µL buffer A and centrifuged at 15,000 r/min for 10 min. Subsequently, the nuclear pellets were resuspended in 25 µL buffer C (20 mM HEPES [pH 7.9], 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM ethylenediaminetetraacetic acid (EDTA), 25% glycerol, 1 mM DTT, 0.5 mM PMSF, and 3.3 µg/mL aprotinin) and incubated at 4℃ for 30 min with occasional vigorous vortexing. The mixtures were then centrifuged at 15,000 r/min for 20 min and the supernatants were used as the nuclear extracts.
Electrophoresis mobility shift assay (EMSA)
The oligonucleotides containing NF-κB and AP-1 binding sites were purchased and used as the DNA probes (Promega) (NF-κB binding site [5′-AGT TGA GGG GAC TTT CCC AGG C-3′] and AP-1 binding site [5′-CGC TTG ATG AGT CAG CCG GAA-3′]). The DNA probes were radiolabeled with [γ-32P]ATP using T4 kinase (Promega). For the binding reaction, the radiolabeled NF-κB and AP-1 probes were incubated with 5 µg of nuclear extracts prepared from the treated cells. In some experiments, a competition study was done with a 100-fold molar excess of unradiolabeled wild-type or mutant NF-κB or AP-1 oligonucleotides. The competitors were added 10 min before the addition of the radiolabeled NF-κB or AP-1 probe. The binding buffer contained 10 mM Tris–HCl (pH 7.5), 50 mM NaCl, 1 mM EDTA, 1 mM DTT, 5% glycerol, and 2 µg poly(dI-dC). The reaction mixture was left at room temperature to allow binding for 20 min. The final reaction mixture was analyzed in a 6.6% nondenaturing polyacrylamide gel with 0.5 × Tris–borate–EDTA as an electrophoresis buffer.
Transient transfections and luciferase assays
We used the transfection reagent TransFast™ (Promega) to transfect plasmids into the cells instead of using electroporation. Briefly, on the day of the transfection, Jurkat T cells were adjusted to 1 × 106 mL−1 in serum-free media and evenly mixed with 2 mg of reporter plasmid (Stratagene, La Jolla, CA) and TransFast™ transfectant (6 mL) in triplicate. Forty-eight hours after transfection, the cells were equally distributed and pretreated or not with various concentrations of amiodarone and then stimulated with or without PMA (5 ng/mL) and ionomycin (1 mM) for 24 h. Subsequently, the cell pellets were collected, the total cell lysates were prepared, and the luciferase activity was determined and assayed according to the manufacturer’s instructions (Promega). Jurkat cells transfected with reporter construct alone were taken as basal activity. Transfections were performed in at least three independent experiments of triplicate determinations. Data are expressed relative to control luciferase activity, normalized to protein, and presented as means +/− SD.
Western blot (Immunoblot) analysis
ECL Western blotting (Amersham, Arlington Heights, IL) was done according to the manufacturer’s instruction and our previous work. 33 Briefly, after extensive washing, the treated and untreated cells were pelleted and resuspended in lysis buffer (20 mM HEPES [pH 7.9], 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride, and 3.3 µg of aprotinin per mL). After periodic vortexing for 1 h, the mixture was centrifuged at 16,000 × g for 20 min, and the supernatant was collected and the protein concentration measured with Lowry assays. Equal amounts (25 µg) of whole cellular extracts were analyzed on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The protein was then transferred to a nitrocellulose filter. For immunoblotting, the nitrocellulose filter was incubated with TBS-T containing 5% nonfat milk (milk buffer) for 1–2 h and then blotted with antisera against p-ERK (extracellular signal-regulated kinase), p-P38, p-JNK (c-Jun NH2-terminal protein kinase) (Calbiochem, La Jolla, CA), IκBα, or beta-actin (PharMingen) overnight at 4℃. After washing with milk buffer twice for 20 min, the filter was incubated with donkey anti-mouse IgG (secondary antibody) conjugated to horseradish peroxidase at concentration of 1:5000 for 1 h. The filter was then incubated with the substrate for 1 min and exposed to an X-ray film.
Statistics
The results were expressed as means ± standard deviation (SD). A paired or unpaired Student’s t-test was used to determine the significance of differences, and a value of P less than 0.05 was considered to be statistically significant. 34
Results
Cytotoxic effects of amiodarone on human T cells
The results of LDH and MTT assays performed on human T lymphocytes indicated that the concentrations of amiodarone used in the experiments were not cytotoxic and did not affect cell viability (Figure 1(a) and (b)).
Amiodarone (Ami) suppressed IL-2, IL-4, IFN-γ, and TNF-α production from activated human T cells. Human T cells at 1 × 106 mL–1 were pretreated with various concentrations of amiodarone (1–5 µM) for 2 h and then stimulated with PMA + ionomycin (P + I) for another 24 h. (a) The supernatants were collected to determine the possible cytotoxic effects of amiodarone with LDH release assays, in which cells treated with 1% Triton X-100 were the positive controls. (b) Cells were incubated with MTT and then analyzed in a microplate reader as described in “Materials and methods” section. (c–f) The supernatants were collected for cytokine measurements. The representative data of at least three to five different donor cells are shown as means±SD. An asterisk (*) denotes statistical significance (p < 0.05) compared with the cells that were stimulated only. EtOH: ethanol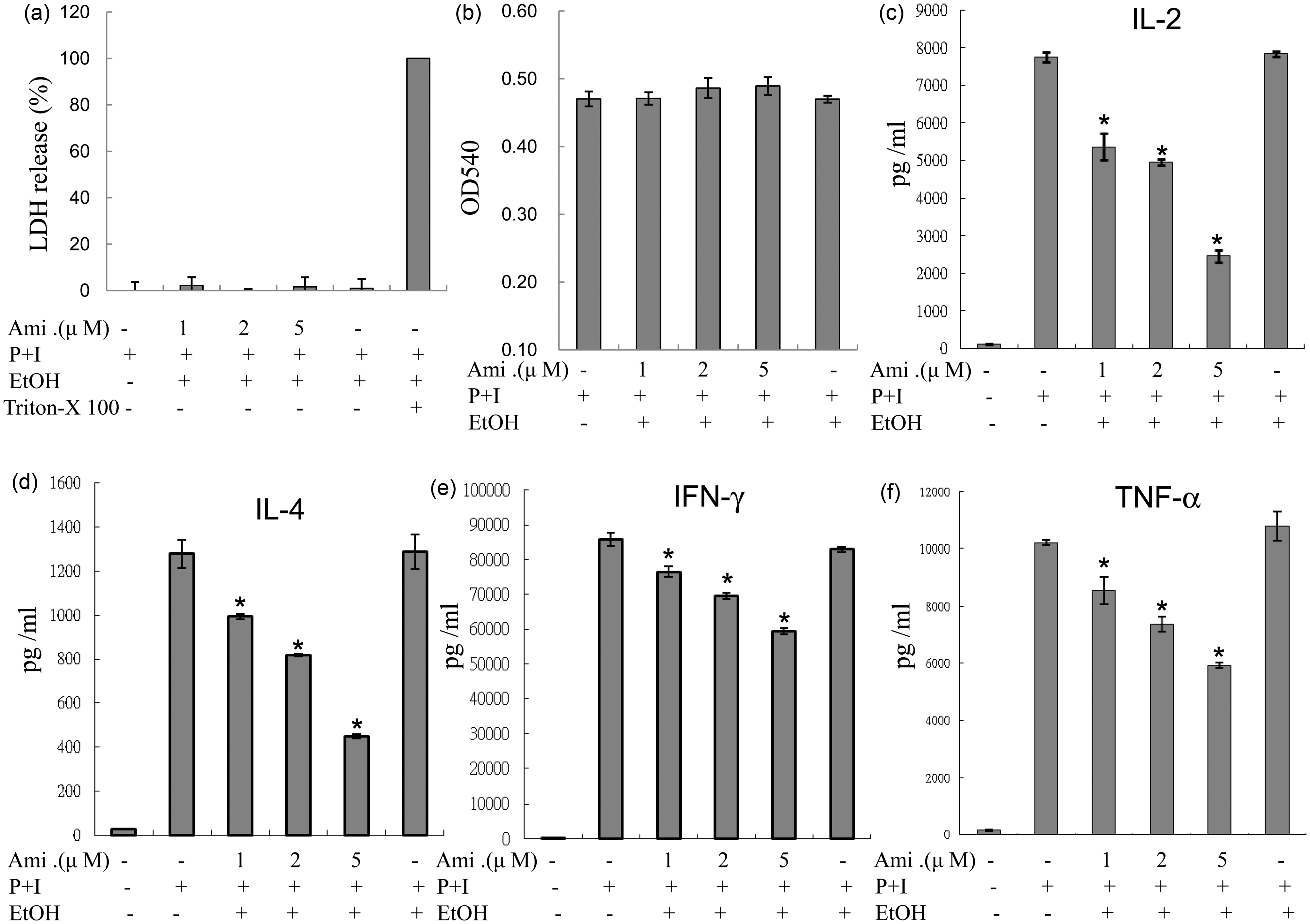
Amiodarone reduced cytokine production from activated human T cells
To examine the effects of amiodarone on the production of cytokines from human peripheral T cells, human T cells were pretreated with amiodarone for 2 h, and then PMA + ionomycin was used to activate human T cells for another 24 h. We found that amiodarone inhibited the stimuli-induced production of cytokines including IL-2, IL-4, IFN-γ, and TNF-α, in human T cells in a dose-dependent manner (Figure 1(c) to (f)).
Amiodarone inhibited T cell activation
Since many T-lymphocyte cytokines were inhibited by amiodarone, we then investigated whether amiodarone affects T cell activation. As shown in Figure 2, the expressions of the cell surface markers of activated T cells, CD25 and CD69, were substantially increased after stimulation with PMA + ionomycin, and this increase was dose dependently decreased by the pretreatment of amiodarone.
Amiodarone (Ami) inhibited the expressions of CD25 and CD69 in activated human T cells. Human peripheral blood T cells at 1 × 106 mL–1 were treated with 1–5 µM amiodarone for 2 h and then stimulated with PMA + ionomycin (P + I) for another 24 h. The expressions of CD25 and CD69 were measured by flow cytometry. The representative data of at least six different donor cells are shown. An asterisk (*) denotes statistical significance (p < 0:05) compared with the cells that were stimulated only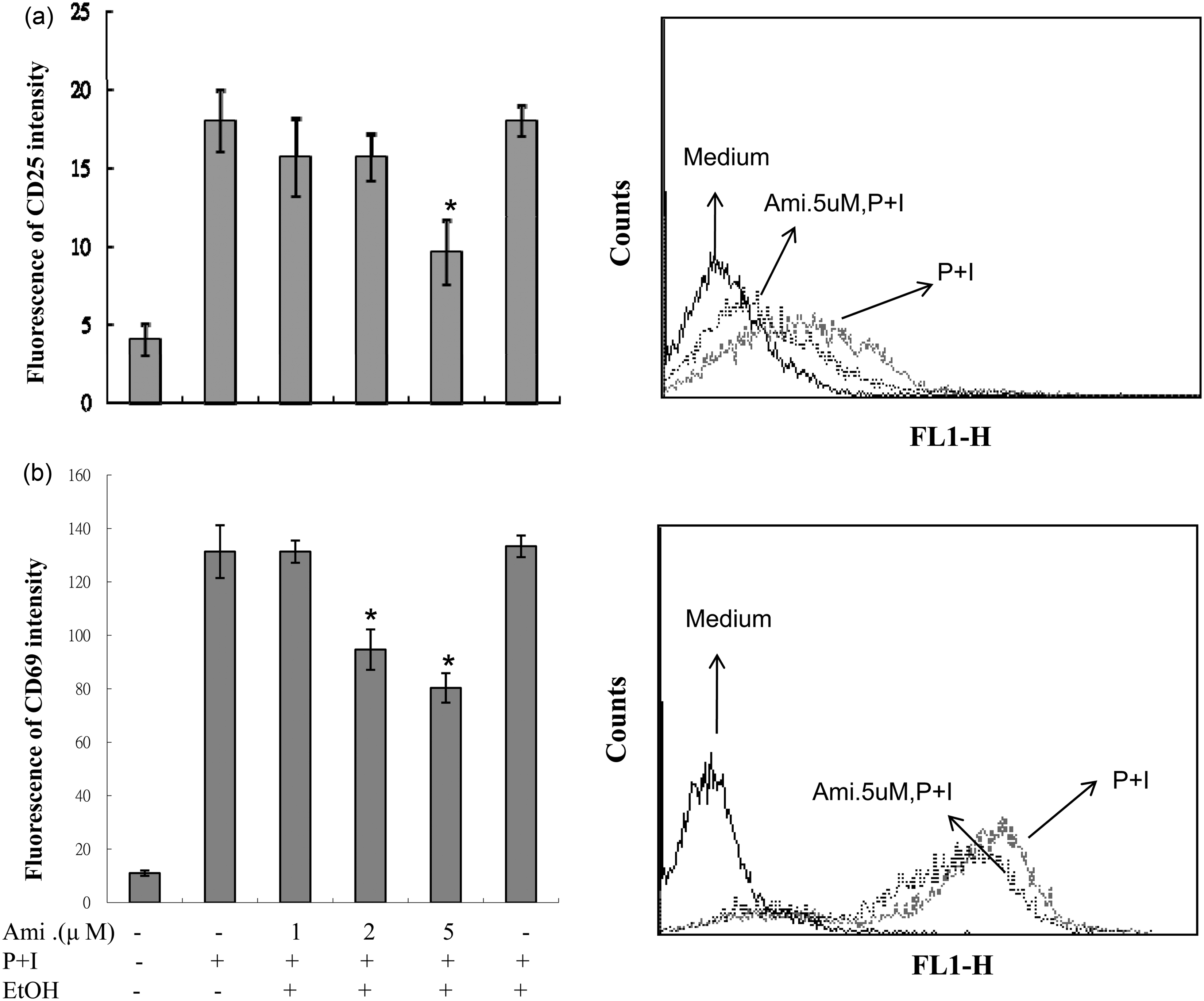
Amiodarone down-regulated AP-1 and NF-κB DNA-binding activities in activated human T cells
Due to the ability of amiodarone to inhibit cytokine production, we hypothesized that amiodarone may modulate some signaling molecules that are commonly involved in T-cell activation. Therefore, we examined the effects of amiodarone on the activation of transcription factors AP-1 and NF-κB. The results showed that amiodarone effectively inhibited AP-1 and NF-κB DNA-binding activities in human T cells activated with PMA + ionomycin (Figure 3(a)). For a more physiological approach, we chose anti-CD3 + anti-CD28 mAbs to stimulate T cells. The results revealed that amiodarone down-regulated AP-1 and NF-κB DNA-binding activities in activated T cells (Figure 3(b)). In competition assays, wild-type AP-1 and NF-κB caused complete disappearance of the band whereas the mutant type did not, indicating the specificity of AP-1 and NF-κB (Figure 3(c)). We further examined whether amiodarone could modulate the AP-1 and NF-κB DNA-binding activities induced by the proinflammatory mediator, TNF-α. As shown in Figure 3(d), amiodarone suppressed TNF-α-induced NF-κB DNA-binding activity, but not AP-1 (data not shown).
Amiodarone (Ami) blocked AP-1 and/or NF-κB DNA-binding activity in human T cells. Human T cells at 2 × 107 mL−1 were pretreated with the indicated concentrations of amiodarone for 2 h and then stimulated with (a) PMA + ionomycin (P + I), or (b) anti-CD3 + anti-CD28 mAbs (CD3 + CD28) for 1 h. Nuclear extracts were then collected. AP-1 and NF-κB DNA-binding activities were determined by EMSA. The
32
P-labeled oligonucleotides containing the NF-κB or AP-1 site were used as probes. The representative data of at least three independent experiments are shown. (c) The specificity of AP-1 or NF-κB binding activity was assessed by competition with the 100 × unlabeled AP-1 or NF-κB as indicated in “Materials and methods” section. Inhibition of the retarded AP-1 or NF-κB band was verified by co-incubation of nuclear extracts with 100-fold molar excess of wild-type AP-1 or NF-κB. In contrast, nuclear extracts co-incubated with mutant-type AP-1 or NF-κB did not affect the band density. (d) Similar to (a) and (b), the effect of amiodarone on NF-κB DNA-binding activity in TNF-α stimulated human T cells was examined by EMSA. The representative data of three different donor cells are shown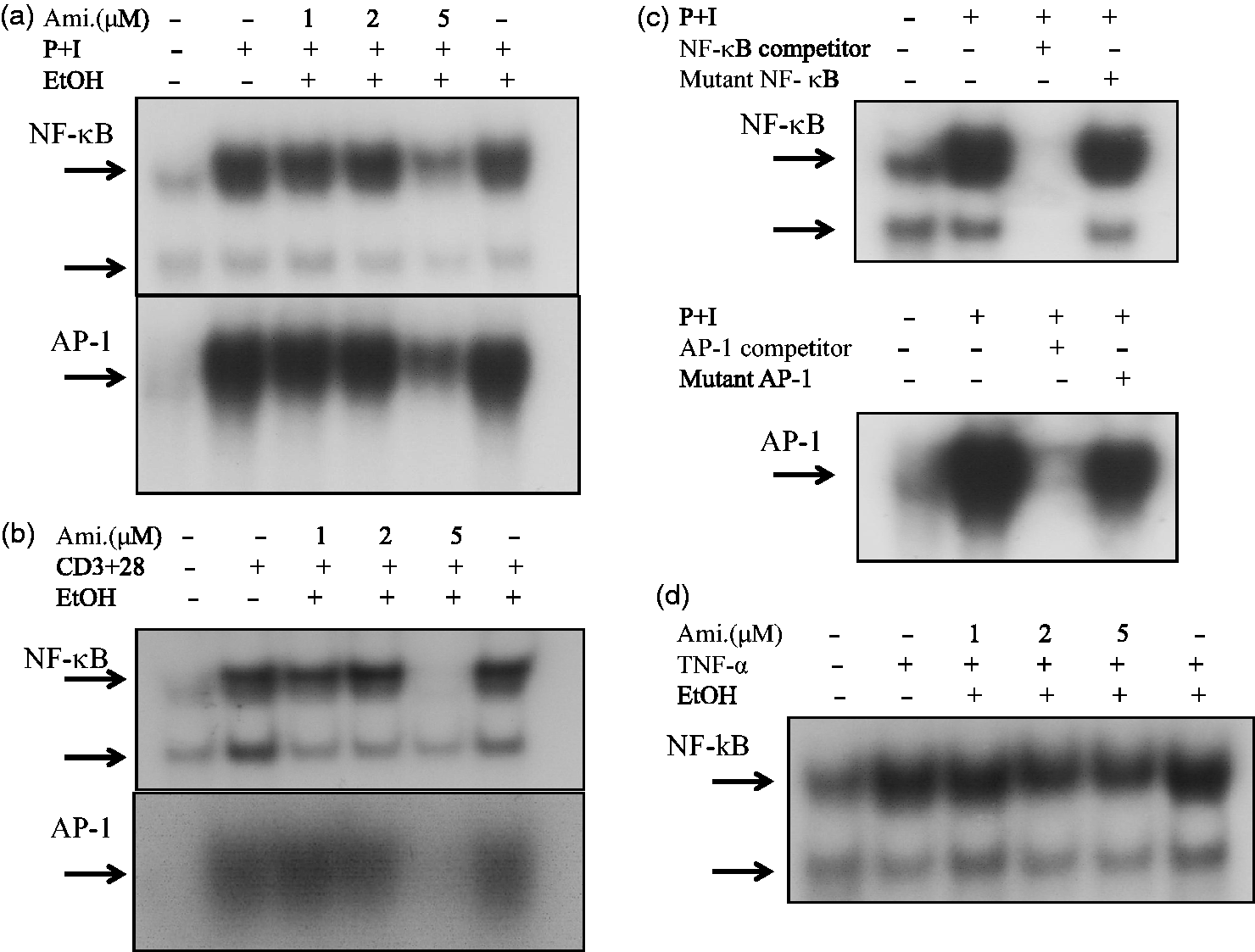
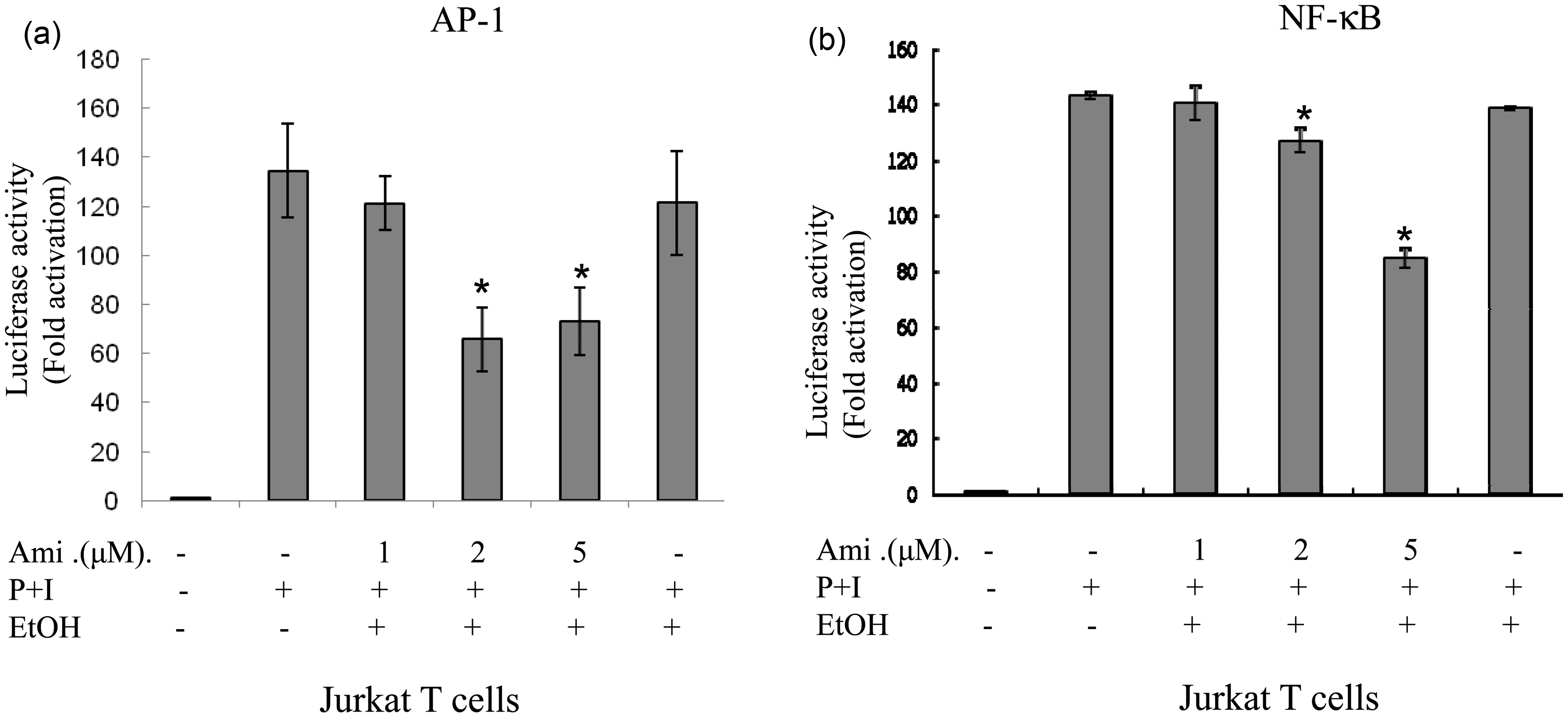
Amiodarone down-regulated AP-1 and NF-κB DNA-binding activities and transcriptional activity in Jurkat cells
To further confirm the effects of amiodarone on AP-1 and NF-κB transcriptional activities, we examined the effects on Jurkat T cells. We transiently transfected AP-1 or NF-κB luciferase reporter plasmids into Jurkat T cells. Forty-eight hours after transfection, the cells were treated with amiodarone at various concentrations and then stimulated with PMA + ionomycin to induce AP-1 and NF-κB transcriptional activity. Consistent with the human primary T-cell observations, amiodarone significantly and dose dependently inhibited both AP-1 and NF-κB DNA binding (Figure 4) and transcriptional activities (Figure 5) in Jurkat cells.
Amiodarone (Ami) inhibited DNA-binding activities of both AP-1 and NF-κB in Jurkat cells. Human T cell line (Jurkat cells) at 2 × 106 mL−1 was pretreated with the indicated concentrations of amiodarone for 2 h and then stimulated with PMA + ionomycin (P + I) for 1 h. Nuclear extracts were then collected. (a) AP-1 or (b) NF-κB DNA-binding activities were determined by EMSA. The representative data of at least three independent experiments are shown Amiodarone (Ami) inhibited transcriptional activities of both AP-1 and NF-κB in Jurkat cells. Human T cell line (Jurkat cells) at 1 × 106 mL−1 was mixed together with (a) pAP-1-Luc (labeled as AP-1) or (b) pNF-κB-Luc (labeled as NF-κB) reporter plasmids and the transfection reagent TransFast™ as described in “Materials and methods” section. At 48 h after transfection, the cells were equally divided and pretreated or not with amiodarone at various dosages for 2 h. After stimulation with PMA + ionomycin for another 24 h, the cells were collected and the total cell lysates were analyzed for luciferase activity. Values are expressed as luciferase activity (fold activation) compared with unstimulated cells. The representative data of at least three independent experiments are shown as means±SD. An asterisk (*) denotes statistical significance (p < 0.05) compared with the cells that were stimulated only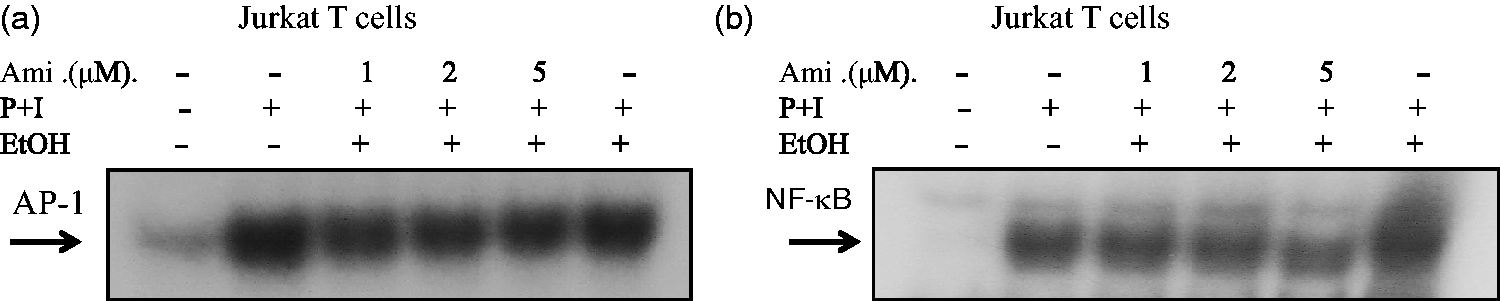
Selective inhibitory effects of amiodarone on JNK and P38 MAPK in activated human T cells
Due to the inhibition of AP-1 DNA-binding activity by amiodarone, we hypothesized whether mitogen-activated protein kinases (MAPKs) would also be affected. Therefore, we evaluated the effects of amiodarone on the activation of MAPK under PMA + ionomycin stimulation. As shown in Figure 6, treatment with amiodarone inhibited the expressions of p-JNK and p-P38 (Figure 6(a) and (b)), but not p-ERK in PMA + ionomycin-activated T cells (Figure 6(c)).
Amiodarone (Ami) inhibited JNK and P38 MAPK signaling pathways and prevented IκBα degradation. Human T cells at 2 × 107 mL−1 were pretreated with the indicated doses of amiodarone for 2 h and then stimulated with PMA + ionomycin (P + I) for another 15–20 min. After washing, the cell pellets were collected and the total cell lysates were analyzed for the protein level. Amiodarone pretreatment suppressed the activity of JNK (a) and P38 MAPK (b), but not ERK (c). Under these conditions, amiodarone had no effect on total JNK, P38, or ERK protein levels. Amiodarone prevented the degradation of IκBα (d). The representative data of four to six experiments are shown. p: phosphorylated; T: total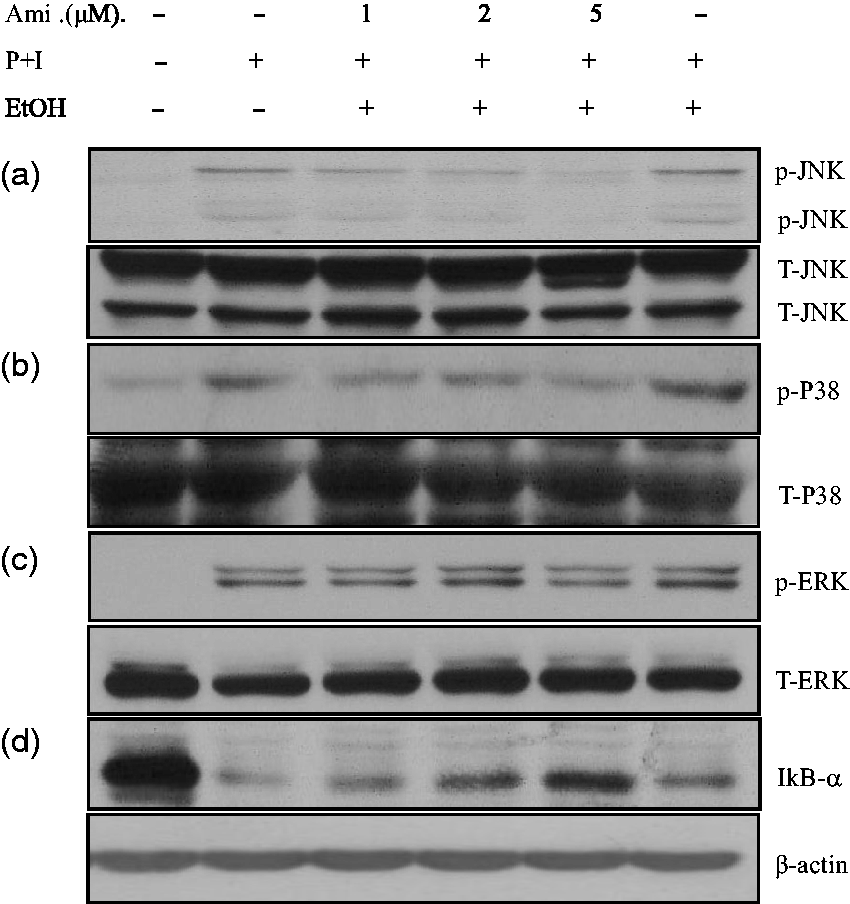
Amiodarone blocked IκBα degradation
The inhibition of NF-κB activity by amiodarone suggested that it may have an effect on the NF-κB-associated protein, IκBα, which retains NF-κB in an inactive status in the cytosol. We further demonstrated that amiodarone significantly suppressed IκBα degradation in PMA + ionomycin-activated T cells (Figure 6(d))
Discussion
Clinically, amiodarone is prescribed for the treatment of Af. One of its therapeutic roles is an anti-inflammatory effect via reducing the activation and movement of neutrophils. 35 Previous studies have suggested that amiodarone alters T cell subsets and regulates T cell functions, which may lead to target organ toxicity such as autoimmune thyroid disease and hypersensitivity pneumonitis in susceptible patients.36,37 In the current study, we showed that one of the beneficial effects of amiodarone is to reduce the activation of stimulated human primary T cells, which may have additional inhibitory effects on immune-related atherogenesis. Further, amiodarone may be able to treat Af in acute coronary syndrome or other clinical conditions related to T cell activation.
The current understanding of the complex mechanisms of Af was summarized in a recent consensus statement published by the Heart Rhythm Society. 38 Although enhanced automaticity, triggered activity, or reentry is thought to induce cardiac arrhythmias, multiple factors from anatomic changes to substrates at the cellular and electrophysiological level also play crucial roles. Among these, inflammation has been highlighted as a risk factor for Af.17,39 As reported in the pathogenesis of postoperative Af, some degree of inflammation and/or oxidative stress may be important. 40 During open heart surgery, the occurrence of Af is related to activation of the complement system, release of proinflammatory cytokines, and increased C-reactive protein levels. Therefore, an inflammatory mechanism in patients with Af has been postulated in light of the high incidence (25–40%) of Af after cardiac surgery.41,42 Furthermore, there is increasing evidence linking inflammation and Af, 43 and this has also been confirmed in several in vivo experiments.44–48 These results suggest that medications inhibiting inflammatory pathways could possibly prevent or treat Af, which further indicates the vital role that inflammation plays in the pathogenesis of Af.
In patients with persistent Af, histopathological evidence points to inflammatory infiltrates as well as increased tissue factor expressions in left atrial appendages. 49 Frustaci et al. reported endomyocardial biopsies of 12 patients with refractory lone Af and found that eight had evidence of inflammation, including lymphomononuclear infiltration and necrosis of the adjacent myocytes in the myocardium. 43 Cytokines are known to be the major mediators in this process. Sata et al. demonstrated that inflammatory markers, including IL-6, TNF-α, and C-reactive protein are elevated in the plasma of patients with paroxysmal Af. 50 In addition, IL-2, secreted by T-helper type 1 lymphocytes, has been reported to be correlated with recent onset Af. 51 IL-2 can also induce the production of IL-6 and activate other cells such as macrophages and vascular cells in the vessels to trigger cytotoxic molecules. 52 Other proinflammatory mediators such as IFN-γ and TNF-α may also trigger detrimental biochemical reactions in chronic cardiovascular events and then induce production of oxygen and nitrogen radicals. Furthermore, IL-4 has been reported to enhance oxidative effects with inflammatory properties.53,54 Therefore, proinflammatory cytokines can be regarded as promoting factors in the progression of Af. Our results demonstrated that amiodarone, an antiarrhythmic agent, could inhibit these inflammatory cytokines and therefore possibly halt the inflammatory pathway of Af.
AP-1 and/or NF-κB could be induced to regulate the gene expression of many cytokines during the inflammatory process. 55 This suggests that the suppressors of cytokine-related signaling pathways may reduce cytokine-induced chronic inflammation. Thus, drug therapy targeted toward minimizing inflammation may be effective in the prevention of Af and its complications. 56 Although several mechanisms are related to the up- and down-regulation of AP-1 activity, MAPKs including ERKs, JNKs, and P38 kinases, are known to be common signaling pathways mediating AP-1 activity.
The activation of NF-κB is considered to be part of a stress response as it is activated by a variety of stimuli. In its inactive form, NF-κB is sequestered in the cytoplasm, bound by members of the IkB family of inhibitor proteins. After cell activation, the IκB proteins are rapidly phosphorylated due to the binding of certain cytokines to their surface receptors. Previous reports have suggested that NF-κB can also be activated in the signaling pathways involved in MAPKs.57,58 Our results demonstrated that amiodarone could suppress AP-1 and NF-κB DNA-binding activity in activated human T cells. Furthermore, we found that amiodarone down-regulated AP-1 and NF-κB transcriptional activity in Jurkat T cells. These results emphasize the anti-inflammatory effects of amiodarone and provide detailed mechanisms of how amiodarone suppresses inflammatory cytokine production.
The limitation of this study is that we only showed ex vivo effects of amiodarone on activated human T cells, and thus making clinical and specific conclusions is beyond the scope of this study. Additionally, the actual role of T cells in atrial arrhythmia during acute coronary events should be further clarified. Nevertheless, our results demonstrate that amiodarone can regulate the processes associated with AP-1 and NF-κB activity in activated human T lymphocytes. In particular, our findings showed that amiodarone exerts these immunomodulatory effects in a therapeutic concentration, which implies that the anti-inflammatory properties of amiodarone will affect T cells in patients with Af.59,60 To clarify these effects, future studies are needed with the inclusion of parallel analysis of T cells from patients who exhibit Af. Moreover, gene expression array analysis in amiodarone-treated human primary T cells as well as Jurkat cells will provide a more complete picture of the anti-inflammatory effects of amiodarone on T cells. This study provides novel concepts regarding the clinical application of amiodarone.
Conclusions
In conclusion, Af is increasingly recognized as a multifactorial disorder, and systemic inflammation may play a significant role in Af. This is the first study to provide data on the potential anti-inflammatory effects of amiodarone on activated primary human T cells. We suggest that amiodarone exerts pleiotropic effects to modulate the function of human T cells and reduce the inflammatory processes of Af. Further in vivo and human clinical studies are needed to elucidate the effects of amiodarone on Af and inflammation-related cardiovascular disorders.
Footnotes
Author contributions
Conceived and designed the experiments: SMC, WHL, CHW, SPY. Performed the experiments: SMC, CSL, LJH, TNT. Analyzed the data: SMC, LJH, JHL, SPY. Wrote the manuscript: SMC, CSL, SPY.
ACKNOWLEDGEMENTS
The authors are grateful for the kind gift (amiodarone) from Sanofi-Synthelabo Pharmaceutical and also gratefully acknowledge Hui-Tzu Tan for his technique assistance. This work was supported by Tri-Service General Hospital grants (TSGH-C98-4-S03 and TSGH-C96-020).