
Abstract
WWOX, the WW domain-containing oxidoreductase gene at chromosome region 16q23.3–q24.1, spanning chromosomal fragile site FRA16D, encodes the 46 kDa Wwox protein, a tumor suppressor that is lost or reduced in expression in a wide variety of cancers, including breast, prostate, ovarian, and lung. The function of Wwox as a tumor suppressor implies that it serves a function in the prevention of carcinogenesis. Indeed, in vitro studies show that Wwox protein interacts with many binding partners to regulate cellular apoptosis, proliferation, and/or maturation. It has been reported that newborn Wwox knockout mice exhibit nascent osteosarcomas while Wwox+/− mice exhibit increased incidence of spontaneous and induced tumors. Furthermore, absence or reduction of Wwox expression in mouse xenograft models results in increased tumorigenesis, which can be rescued by Wwox re-expression, though there is not universal agreement among investigators regarding the role of Wwox loss in these experimental models. Despite this proposed tumor suppressor function, the overlap of the human WWOX locus with FRA16D sensitizes the gene to protein-inactivating deletions caused by replication stress. The high frequency of deletions within the WWOX locus in cancers of various types, without the hallmark protein inactivation-associated mutations of “classical” tumor suppressors, has led to the proposal that WWOX deletions in cancers are passenger events that occur in early cancer progenitor cells due to fragility of the genetic locus, rather than driver events which provide the cancer cell a selective advantage. Recently, a proposed epigenetic cause of chromosomal fragility has suggested a novel mechanism for early fragile site instability and has implications regarding the involvement of tumor suppressor genes at chromosomal fragile sites in cancer. In this review, we provide an overview of the evidence for WWOX as a tumor suppressor gene and put this into the context of fragility associated with the FRA16D locus.
Introduction
WWOX, WW domain containing oxidoreductase, is a large gene (1.2 Mb open reading frame) with a relatively small, 2.2 kb transcript.
1
A schematic of WWOX is denoted in Figure 1(b) showing nine exons and a particularly large eighth intron spanning 779,639 bp.
2
The Wwox protein contains two WW binding domains at its N terminal region and a short-chain dehydrogenase/reductase (SDR) domain in its central region.
1
Characterization of these domains has been an important part of describing the cellular and physiological functions of Wwox.
Chromosome fragility and genetic alterations at the WWOX/FRA16D locus. (a) Metaphase spread showing G-banded metaphase chromosomes and homozygous breaks at 16q23, with corresponding chromosome 16 ideogram, identifying the colocalization of WWOX and FRA16D. (b) Schematic of the WWOX gene showing exons as white boxes below their corresponding genomic location. Genetic alterations are listed in blue (homozygous) or orange (heterozygous) with deletions denoted as gaps. Samples shown for homozygous deletions are epithelial cancer cell lines: PEO1/4/6 cell lines derived from ovarian adenocarcinoma, SCLC derived from small cell lung carcinoma including the cell lines WX330 and NCI-H69, PANC1 cell line derived from pancreatic ductal adenocarcinoma, HCT116 cell line derived from colorectal carcinoma, AGS cell line derived from stomach adenocarcinoma, CO-115 and KM12C cell lines were derived from colon carcinomas. Heterozygous deletions were detected in breast and prostate cancers via PCR microsatellite analysis. (c) Replication-sequencing data generated from ENCODE for the WWOX locus in epithelial cells. Cell cycle phases are indicated on the left with S phase subdivided into four fractions. The WWOX locus relies on long-traveling forks (gray arrows) emanating from replication origins (orange circles) located in the flanking regions to converge in G2 phase, resulting in late completion of replication for the center of the gene. (A color version of this figure is available in the online journal.)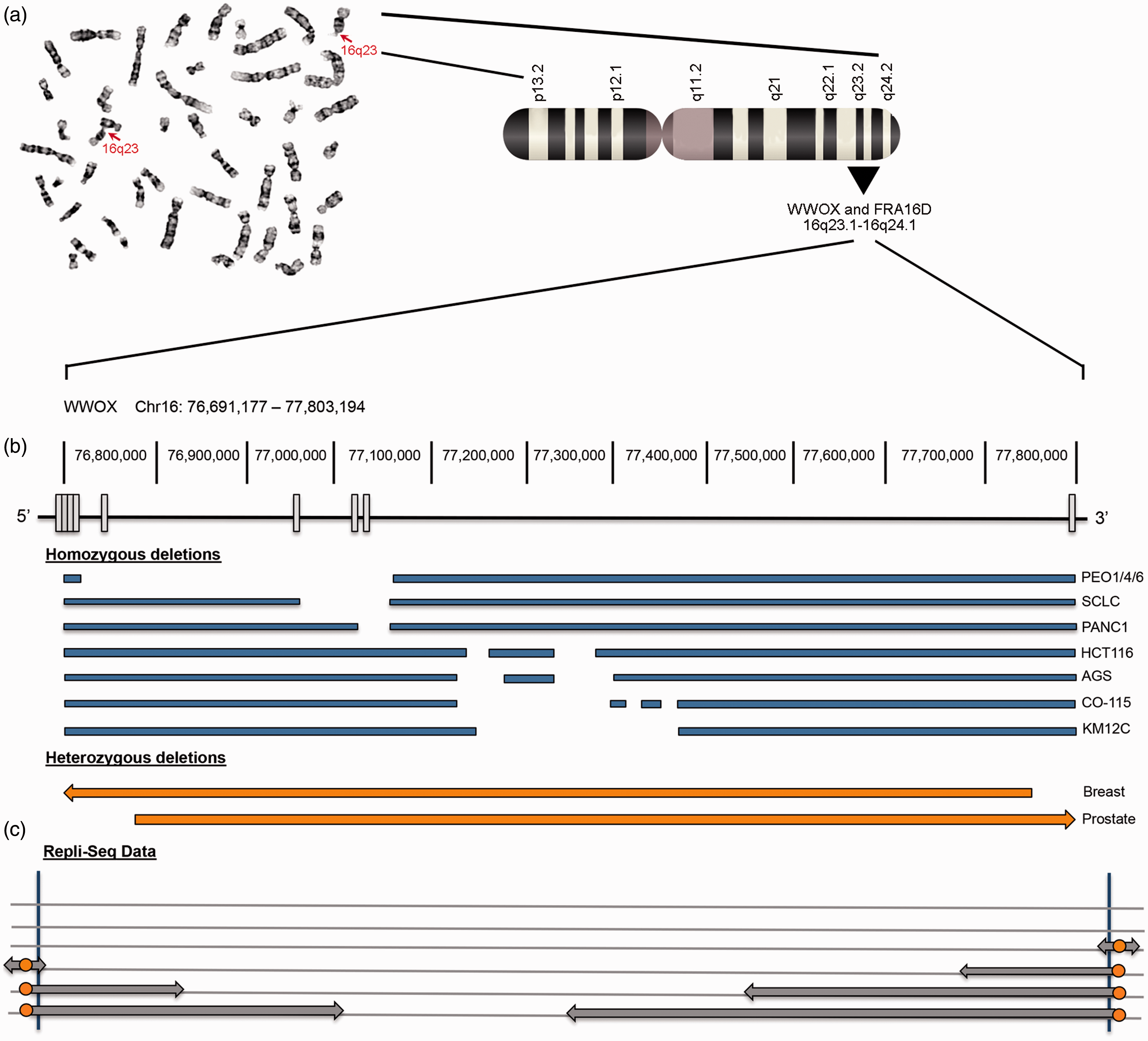
WW domains are small protein modules named for their unique structure: two conserved tryptophan (W) residues spaced approximately 20 amino acids apart. 3 Functionally, WW domains are recognized for their involvement in protein–protein interactions and grouped according to their binding preference to proline-rich ligands. Initially, Wwox was thought to belong to Group I after demonstrating binding of proteins harboring PPxY motifs (where P is proline, Y is tyrosine, and x is any amino acid) through its first WW domain.4–6 However, a recent study by Abu-Odeh et al. employed an MS-based screen which confirmed previous PPxY protein interactions, but also demonstrated that the majority of Wwox interacting proteins did not contain PY motifs. 7 Rather, these proteins exhibited PPxF or LPxF motifs (where F is phenylalanine and L is leucine), suggesting that perhaps WW1 domain of WWOX binds non-canonical proline-rich motifs. Many studies have shown that WW1 domain interacting proteins do not interact with the WW2 domain.5–7 After showing that the second WW domain, WW2, contains two distinct amino acid residues within the WW binding pocket, compared to WW1, McDonald et al. proposed that the WW2 domain serves as a chaperone for augmenting physiological binding of WW1. 4
When the WWOX gene was cloned in 2000, Bednarek et al. predicted that the SDR domain enabled Wwox to play a role in steroid metabolism. 1 Seven years later, a Wwox knockout mouse was generated by Aqeilan et al. and found to succumb postnatally to a severe metabolic syndrome. 8 The phenotype of this mouse was carefully characterized through immunohistochemistry and affymetrix gene expression analysis to demonstrate that Wwox plays critical functions in both gonadal development and the steroidogenesis pathway. 9 Subsequent lipoprotein profiles in liver-specific Wwox knockout mice have suggested a role for the protein in cholesterol homeostasis and fatty acid biosynthesis/triglyceride metabolism. In support of this proposed role of Wwox in lipoprotein and steroid metabolism, Iatan et al. have characterized variants within the WWOX gene which segregate with dyslipidemia in two French Canadian families. 10 Interestingly, WWOX expression levels are highest in hormonally regulated tissues such as the ovary, prostate, and testes. 1 Thus, Wwox protein appears to play critical roles in lipoprotein, high-density lipoprotein, and sex steroid metabolism, although the contribution of the SDR domain to this phenotype remains to be confirmed.
The WWOX gene also spans the common chromosomal fragile site (CFS) FRA16D. The term “fragile site” was first used to describe gaps that appeared on chromosomes in culture following replication stress. 11 Initially, replication stress was induced by treatments of folate deprivation or dihydrofolate reductase inhibition, 11 but the mild polymerase inhibitor, aphidicolin, is now traditionally used to precipitate CFSs. 12 Fragile sites gained attention when the rare fragile site FRAXA, located at Xq27, was associated with X-linked mental retardation, also known as fragile X syndrome. 13 Common fragile sites were identified when a group of different, recurring fragile loci were observed in control samples when studying FRAXA. 12 Fragile sites are classified as rare or common depending on their frequency within the population. 14 CFSs are identifiable in all individuals, while rare fragile sites are seen in less than 5% of the population. Rare fragile sites segregate in a Mendelian manner and exhibit nucleotide expansions, such as the CCG trinucleotide repeat that characterizes FRAXA. 15 In general, CFSs harbor large transcriptionally active genes, usually greater than 1 Mb in length. 16 The two most frequently activated and well-studied CFSs in lymphocytes are FRA3B at 3p14.2, encoding FHIT and FRA16D encoding WWOX. 17
In addition to being located at fragile sites, FHIT and WWOX have also both been reported to be tumor suppressor genes,8,18 whose loss is associated with numerous human cancers.19,20 In 1979, the first chromosomal translocation associated with familial cancer, a renal cell carcinoma, was mapped to a breakpoint in 3p21. 21 Following the identification of CFSs in 1984, scientists quickly noticed that the non-random alterations associated with cancer frequently coincided with the location of CFSs. 22 Accordingly, the 3p21 breakpoint was later hypothesized to be near FRA3B at 3p14.2 23 and the FHIT gene was cloned at 3p14.2 in 1996, 24 showing the overlap of FHIT and FRA3B loci. In parallel to the search for CFSs, cancer researchers were attempting to map tumor suppressor genes by homozygosity mapping. In the 1990s loss of heterozygosity (LOH) and allelic imbalances in breast cancers were mapped to the long arm of chromosome 16, 25 the location of FRA16D and WWOX. 1
Two opposing views dominate the discussion regarding the role of CFSs in cancer. One school of thought is that genomic instability created by cancer progression causes collateral damage to FHIT and WWOX. In this way, they are thought to be unselected “passenger” mutations in cancers. 26 The counterargument is that deletions and other genomic alterations at FHIT and WWOX loci at CFSs occur early in cancer initiation or progression, and cancer cells with clonally unique FHIT or WWOX gene deletions are selectively expanded due to loss of tumor suppressor functions such as protection of genome stability 27 or programmed apoptosis.28,29 In this review, we discuss the arguments for WWOX as a tumor suppressor and put this into context of recent discoveries regarding CFSs.
Chromosomal fragility
Understanding the mechanism underlying chromosomal fragility is an important precursor to determining the biological effects of gene deletions at fragile sites. Until three years ago, the prevailing explanation for chromosomal fragility was the presence of sequences affecting replication fork dynamics. The replication fork is generated when the helicase/topoisomerase complex travels just upstream of polymerase to uncoil and separate the DNA strands. With aphidicolin-mediated replication stress, the helicase proceeds uncoupled from DNA polymerase, generating long stretches of single-stranded DNA. Nucleotide repeats, associated with rare fragile sites, were believed to form secondary structures within these stretches of single-stranded DNA and impair movement of the replication fork, causing its collapse and the DNA breaks seen cytogenetically as chromosomal breaks. 16 Since rare, heritable fragile sites were discovered to involve expansion of nucleotide repeats, it was logical to suppose that common fragile sites also owed their fragility to the actual nucleotide sequence. As these sequences were discovered and reported, efforts were made to discover sequence elements within them that could account for their fragility. A theory that was widely investigated was put forward by the Kerem laboratory, a proposal that flexible regions of DNA within CFS loci could cause secondary structures that could interfere with replication fork progression under conditions of mild replication stress. 30 However, it was difficult to identify sequences that would generate predictable secondary structures in single-stranded DNA for common fragile sites that would be true only of fragile sites.17,31 For instance, the FRA3B and FRA16D loci are AT rich compared to the rest of the genome, but this in itself cannot explain fragility, as comparable AT enriched genomic regions are not fragile. Other theories proposed that differences in chromatin and replication-associated proteins at the fork were responsible for differences in susceptibility to DNA breaks in fragile versus non-fragile regions. 32
In 2011, the laboratory of Michelle Debatisse investigated epigenetic mechanisms associated with FRA3B. 33 Letessier et al. used DNA combing experiments, where stretched-out DNA strands are pulse labeled with fluorescent tags in vivo during replication, to evaluate replication fork speed and symmetry. Surprisingly, the authors found that replication forks were not stalling along the FHIT locus. They employed replication-sequencing (repli-seq) data to evaluate the distribution of replication initiation and termination events at FRA3B and showed that in lymphoblasts (the white blood cell type previously used to identify and describe the frequency of CFS) the FRA3B locus has no replication initiation origin within a central 700 kb region of the 1.2 Mb gene. This is a significant scarcity of replication origins since other genomic regions of similar size have approximately 10 replication origins. With no central replication origins, replication forks from flanking regions of the locus must cover very long distances (10 times longer than normal) to converge and complete replication. If a replication stressor such as aphidicolin is applied to lymphoblasts, replication at loci like FRA3B may not reach completion before entering G2 and mitosis phases, causing chromosomal gaps in mitosis, the defining feature of CFSs. Letessier et al. also examined repli-seq data for FRA3B in fibroblasts, cells derived from connective tissue. Fibroblasts exhibited a different replication origin pattern that corresponded to the lack of fragility at 3p14.2 in fibroblasts compared to lymphoblasts. In this way, Letessier et al. showed that different cell types exhibit different common fragile sites based on an epigenetically determined feature, the placement of replication origins across genomic loci in cells of specific tissue origins. 33
Genetic alterations and fragility of Wwox
If fragility of FRA16D is indeed due to unfinished replication caused by a scarcity of replication origins, a review of the published deletion locations of WWOX should correspond to those regions prone to breakage. Genetic alterations at the WWOX locus occur through LOH, homozygous deletions, and translocations. 20 LOH, or loss of one allele or a portion of one allele of the WWOX gene, has been reported for 52% of hepatocellular carcinomas, 53% of prostate carcinomas, 67% of breast carcinomas as well as for lung and gastric cancer.34–36 Homozygous deletions between exons 4 and 9 have been characterized in colon, ovarian, small cell lung, and pancreatic cancer cell lines.34,37 Translocations have been reported in multiple myeloma 38 and the colon cancer cell line, HCT116. 39 Likewise, aberrant transcripts have been reported for Wwox,40–42 also showing loss of exons 4 and 8 in the majority of cases, indicating a similar pattern for these exon deletions. Figure 1 shows examples of homozygous deletions and LOH that have been documented for WWOX in several cancer types and cell lines. Most losses occur between exons 4 and 8, the region that corresponds to the late-replicating region by repli-seq data. Altogether, the Debatisse theory of fragility supports the genetic alteration patterns thus far reported for WWOX.
Since fragility studies were initially conducted in lymphoblasts, 12 identifying the cell-type specific mechanism of fragility has prompted a re-evaluation of CFSs in different cell types. As such, Hosseini et al. evaluated cells from epithelial tissues, the most common tissue origin for human cancers, for location and frequency of common fragile sites. 43 The authors used epithelial cell lines from breast, colorectal, and bronchial epithelial tissue to evaluate common fragile sites from metaphase spreads in order to appropriately provide a new CFS ranking for epithelial cells. They also evaluated repli-seq data provided by publicly available databases (ENCODE) in order to put their results into context with the epigenetic theory of fragility proposed by the Debatisse laboratory. Cytogenetic analyses demonstrated that the most fragile locus in all epithelial cells examined, with the exception of the bronchial epithelial cell line, was FRA16D rather than FRA3B. This implies that FRA16D is the most fragile locus in most epithelial-derived human cancers. In a similar study, Le Tallec et al. analyzed epithelial (colorectal and breast) and erythroid-derived cancer cell lines to identify CFSs and quantify fragility in these cell types. 44 FRA16D was the only fragile site identified in all four cell types compared (lymphoblasts, fibroblasts, epithelial, and erythroid cells) and was consistently one of the top three CFSs for epithelial cells, showing more fragility than FRA3B in all cell lines except one. 44 This is a significant finding as FRA3B has overshadowed FRA16D for nearly three decades as the “most fragile site.” It would then follow that fragility of different tumor suppressor genes should be put into context of the cell type from which the tumor originates.
Putative Wwox biological functions
In order to determine the relevance of FRA16D fragility and resulting WWOX deficiency in cancers, a review of recent functional studies is required to determine putative roles of Wwox in tumor suppression. The function of Wwox can be defined, in part, by the company of the partners its binds. A growing list of Wwox interacting proteins have been described and include transcription factors p73, 45 Ap2γ and γ, 46 Jun, 47 Runx2, 48 and PPxY independent interactions with p53 and Mdm2. 49 These transcription factors regulate pathways involving cellular apoptosis, proliferation, and development. 50 Other Wwox interacting proteins of notable physiological relevance include: ErbB4, 5 a receptor tyrosine kinase involved in mitogenesis and differentiation, and Dvl-2, 51 a stabilizing protein involved in the non-canonical Wnt/β-catenin pathway which elicits cell growth and proliferation. Not only does Wwox serve a variety of functions in distinct intracellular pathways contingent on the identity of its binding partner, Gourley et al. also show that Wwox modulates interactions between tumor cells and the extracellular matrix (ECM). 28 Through in vitro and in vivo studies involving the transfection of WWOX into ovarian cancer cell lines, the authors showed that adhesion of tumor cells to fibronectin in the ECM is dependent on WWOX expression via membrane integrin α3. 28 This suggests that Wwox serves important, yet diverse intracellular and extracellular functions.
Wwox cellular localization also provides some clues to its function. Most studies report Wwox protein localized to the cytoplasm where it sequesters transcription factors (Ap2γ and p73) 46 or signaling proteins such as Dvl-2, to prevent nuclear import and transactivation or Wnt pathway activity.5,51 In this way, Wwox deficiency could result in the deregulation of numerous physiologically relevant pathways through binding and repression of a variety of transcription factors and signaling proteins in the cytoplasm. Other reports have suggested that Wwox localizes to the golgi apparatus, 2 as well as the nucleus and mitochondria.49,52 With the large array of binding partners identified, it is possible that Wwox participates in pathways located in distinct compartments within the cell. The case for definition of Wwox function becomes more complex in some instances, as with ErbB4 and Wwox binding in breast cancer cells. 5 Here, Wwox competes for ErbB4 intracellular domain binding, with other WW domain containing proteins, Yap and Itch, involved in the same pathway but producing opposing downstream effects. 5 Although studies are beginning to shed light on the myriad functions of Wwox, it is clear that much work is to be done in order to characterize the full extent and particular mechanisms in which Wwox deficiency may potentiate tumor cells. It will be necessary to learn much more about which WW domain containing proteins are expressed in specific tissues or niches where Wwox is expressed, and how these expression patterns change with development of cancer and progressive stages of cancer, in order to follow the signal pathways directed by Wwox within the complex WW domain network of signaling proteins.
Wwox deficiency in cancers
Evidence for WWOX as a tumor suppressor gene began with reports that a variety of epithelial cancers, including breast carcinomas, preinvasive breast lesions, prostate and hepatic carcinomas exhibited LOH at 16q.53,54 Several efforts then culminated in cloning of the WWOX gene and identifying its overlap with FRA16D on the long arm of chromosome 16.1,40 Since then, studies have confirmed that WWOX expression is decreased or absent in many human cancers, reviewed in Gardenswartz and Aqeilan
20
and highlighted in Figure 2. Interestingly, Wwox deficiency is often correlated with breast, prostate, and ovarian cancers,55,56 tissues for which WWOX expression is highest. Studies have demonstrated the important physiological role that Wwox plays in mammary gland ductal development as knockout mice exhibit impaired ductal growth and increased fibronectin levels.
57
There is also evidence of significant Wwox deficiency in human invasive breast carcinoma tissue samples
58
and breast cancer cell lines.
42
Furthermore, reduced WWOX expression is highly correlated with various clinicopathologic factors such as triple negative breast cancers,
59
basal phenotypes,
60
and tamoxifen resistance.
61
WWOX/Wwox expression alterations in human cancers. Summary of reports showing correlations between reduced/absent WWOX/Wwox expression, LOH (loss of heterozygosity), or normal WWOX expression in various common human cancers. (A color version of this figure is available in the online journal.)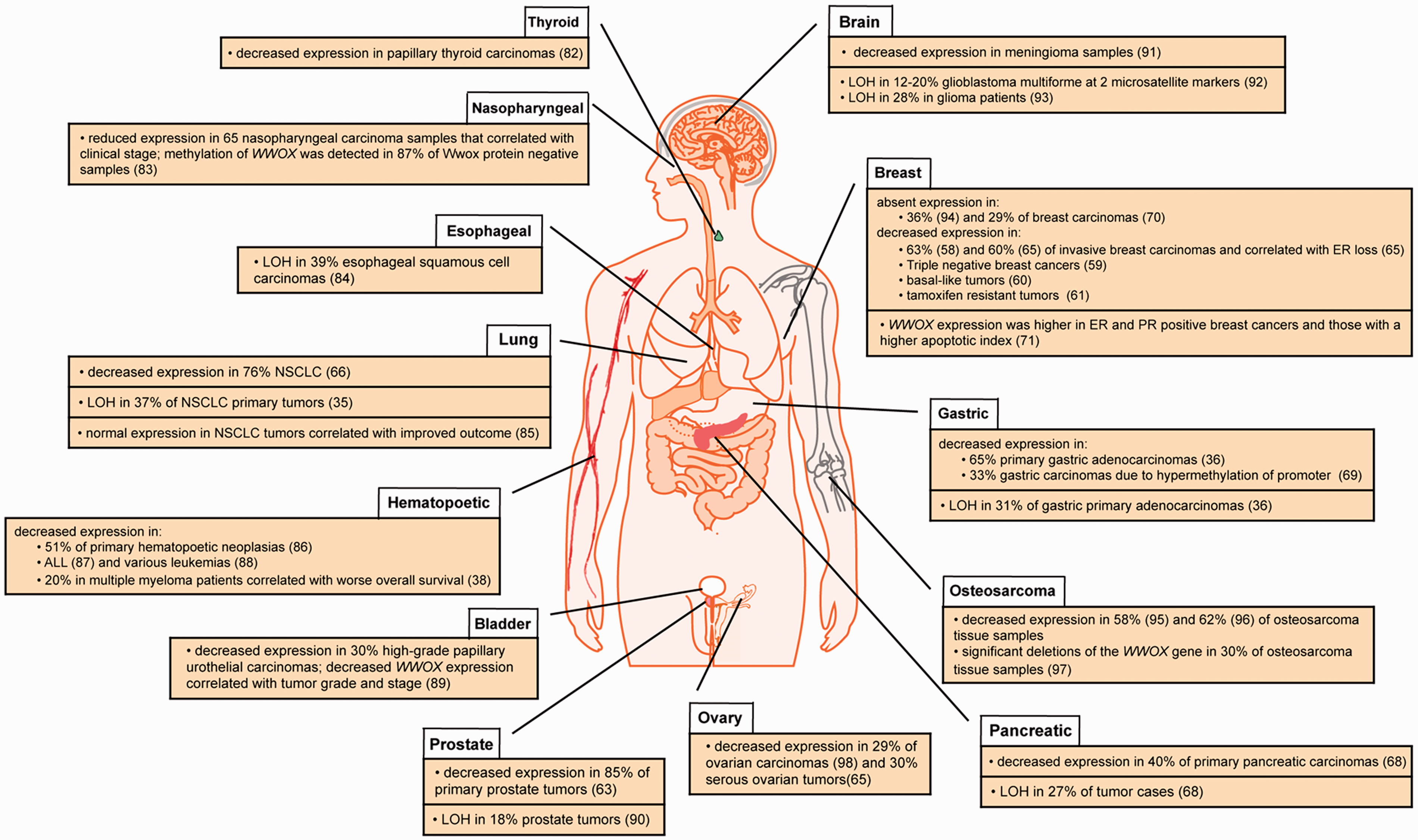
In terms of prostate cancer, WWOX has been identified as an area of genomic loss in tumors based on aCGH 62 and its expression has been found to be decreased in 84% of tumors analyzed via immunohistochemistry. 63 Furthermore, Qin et al. found that WWOX overexpression in vitro induced apoptosis and stalled cell growth, suggesting a mechanism for WWOX tumor suppressor function in prostate cancer cells. As mentioned earlier, a similar mechanism for the involvement of WWOX in ovarian cancers has also been proposed. 28 Accordingly, decreased WWOX expression has been significantly correlated with advanced clinical stage (FIGO Stage IV), decreased overall survival, and lymph node metastasis in ovarian cancers.64,65
WWOX deregulation has also been associated with a number of non-hormonally regulated cancers such as lung, pancreatic, gastric, bone, and skin cancers, as well as others highlighted in Figure 2. Non-small cell lung tumor samples were found to exhibit decreased WWOX expression,35,66 which correlated with certain histotypes as well as increased tumor aggressiveness. 67 Pancreatic and gastric cancers also demonstrated WWOX LOH as well as decreased expression in primary tumors and cell lines.36,68 Furthermore, WWOX transfection inhibited pancreatic cell colony formation via apoptosis. 68 In summary, WWOX expression is frequently decreased in a wide variety of cancers. In some cases, WWOX plays a key developmental function (mammary ducts) but more often it is involved in promoting apoptosis, as in the case of detached ovarian and prostate cancer cells, revealing its tumor suppressive function.
WWOX: a non-canonical tumor suppressor
Watanabe et al. challenged the notion of WWOX as a tumor suppressor due to their finding that tissues from 10 of out of 16 gastric tumors and five out of five breast tumors exhibited normal or increased WWOX expression. 52 Subsequent studies evaluating many more tumor samples and in many more cancers have demonstrated a significant correlation with decreased WWOX expression in gastric cancers36,69 and breast cancers,58,65,70 sometimes also correlating with more aggressive disease and worse prognosis.59,61,71 It is also possible that losses or changes in the balance of expression of specific Wwox-binding or competing WW-domain proteins could lead in some cases to gain of WWOX expression.
Initial studies of WWOX also described a lack of point mutations along the gene length, a classic sign of a tumor suppressor. 1 A recent examination of breast cancer tissue samples identified 13 point mutations in 81 breast cancer tissue samples. 72 One mutation was considered a previously reported polymorphism, 64 however of the remaining 12, four were categorized as non-sense mutations and eight as missense mutations in the coding sequence. Still, numerous reports failed to find non-polymorphic point mutations within Wwox1,64,66,68,73 suggesting a need for confirmation of these findings. We propose that due to its location at a CFS, the WWOX locus is so sensitive to damage-induced chromosomal gaps or breaks at late-replicating sites within this large gene, that LOH and homozygous deletions occur at a much higher frequency than might be expected for occurrence of point mutations. In addition, WWOX silencing occurs via other classical tumor suppressor mechanisms such as hypermethylation of promoter regions and ubiquitination in breast, lung, bladder, and prostate cancers.74,75
Interestingly, the overlap of putative tumor suppressor genes and CFSs is highly conserved over many species, including mice.76,77 Mouse models have confirmed the tumor suppressive function of the WWOX ortholog. Wwox+/− mice have a significantly higher incidence of spontaneous lung and mammary tumors and are more sensitive to DNA damaging agents compared to wild-type mice.45,46 Significantly, some of these tumors from Wwox+/− mice retained one intact Wwox allele, suggesting haploinsufficiency as a mechanism for loss of tumor suppressive function. Complete knockout mice exhibit osteosarcomas by 3–4 weeks of age 8 and Wwox hypomorphic mice display spontaneous B-cell lymphomas. 78 On the other hand, although the Wwox+/− mouse on a C3H mammary tumor-susceptible genetic background exhibited enhanced mammary tumorigenesis, 79 mice with targeted deletions of Wwox in mammary tissue did not show a higher incidence of mammary tumors.57,80 These conflicting results could be explained by the varying impact of ECM interactions with cancers—as Wwox was decreased in all tissues in the C3H mouse, but only in epithelial mammary cells in the conditional knockouts. In this way, the interactions with the ECM, such as that proposed by Gourley et al. in ovarian caner cells, 28 could contribute to its tumor suppressor function in mammary cancers. It is also possible that loss of Wwox contributes to the progression of mammary carcinogenesis rather than the initiation.
Despite much evidence suggesting an association of WWOX deficiency in various cancers and the selective advantage it affords cancer cells, speculation continues regarding the role of tumor suppressor genes located at CFSs, such as WWOX and FHIT, in carcinogenesis. Proponents of the passenger theory, regard homozygous deletions, a hallmark of tumor suppressor genes in cancer, as bystander effects of WWOX and FHIT fragility. 26 Taken together with the replication origin pattern of fragility, one would predict that reduced WWOX expression would be correlated with nearly all epithelial cancers, since it is the most fragile locus in epithelial cells. However, McAvoy et al. determined the frequency of fragile sites for various cancers and then compared the expression of corresponding genes located at their CFS loci. 81 Interestingly, there was no correlation between fragility and the frequency with which the corresponding genes were deleted. Furthermore, some cancers did not have inactivation of any of the large genes located at CFSs, such as WWOX, suggesting specific selection criteria for CFS gene inactivation in different cancers. Recently, Le Tallec et al. demonstrated through elegant and detailed erythroid and epithelial cell CFS profiling that over 50% of recurrent cancer deletions originate from CFSs associated with large genes, demonstrating new significance for the contribution of genes located at CFSs in cancer. 44
Conclusion
Due to its location at a CFS, its lack of point mutations, and conflicting mouse mammary cancer model data, the function of WWOX as a tumor suppressor gene has been challenged. We have reviewed data showing significantly decreased expression of WWOX in various human cancers, in some cases correlating with histotype and prognosis. In addition, there are numerous studies that imply involvement of WWOX in apoptosis, cell growth, and/or proliferation, all functions that contribute to a tumor suppressor role.
In light of the recent findings highlighting the exquisite fragility of FRA16D, we propose that replication stress caused by the underlying genomic instability of cancer cells results in deletions at WWOX due to its overlap with the FRA16D. This mechanism of alteration to expression could explain or contribute to the scarcity of point mutations found within the WWOX gene. The presence of WWOX in clonal populations of heterogenous cancer cells suggests that following FRA16D-associated chromosome rearrangements, WWOX deficiency may provide a selective advantage for these cells—possibly through tissue- and cell-specific interactions with intracellular and extracellular proteins expressed in the particular cancer tissues. For example, in prostate tissues, the binding of WWOX with the transcription factor Ap2γ prevents Ap2γ activity in the nucleus and subsequent activation of ERBB2, a mediator of androgen receptor activity and prostate cancer cell growth. 63 In a Wwox-deficient state, Ap2γ is free to activate ERBB2 and promote cancer cell growth. However, in other tissues, which lack ERBB2 and androgen receptors, loss of Wwox might not provide a growth advantage and so not promote the clonal expansion of those cells; therefore, Wwox deficiency would not be associated with cancers in those tissue types. This idea is in accord with the findings by McAvoy et al. that WWOX deficiency was not found in all cancer cells where WWOX/FRA16D was the most fragile CFS—perhaps it is only found at high frequency in tissues in which its myriad protein–protein interactions enable it to provide a selective advantage. One could speculate that with the many putative binding partners and pathways associated with Wwox, and the array of complex inter-relationships among them, such as those involving Wwox, ErbB4, Yap, and Itch, that many possible scenarios complicate elucidation of a Wwox tumor suppressor role in mouse mammary tumor models.
The proposal by Le Tallec et al. that over 50% of deletions in human cancers occur at CFSs emphasizes the importance of defining specific sites of chromosome fragility in different cancer types and understanding the biological context and function of genes located at those sites. The discoveries regarding CFSs have highlighted the importance of Wwox owing to its location at FRA16D, the most frequent fragile site in epithelial cells, the most common origin of human cancers. More research is necessary to understand the biological and tumor suppressor functions of WWOX. It is necessary to understand when WWOX loss occurs in cancer progression but also to continue illuminating the various functions of Wwox protein in distinct tissue types to more fully characterize its tumor suppressor function.
Footnotes
Authors’ Contributions
MSS wrote the manuscript. KH edited and provided helpful discussions.
Acknowledgements
We thank Teresa Druck for figure preparation and Huebner lab members for constructive feedback. This research has been supported by 9T32 OD010429, U01 CA154200, and R01 CA120516.