
Abstract
Abnormal mitochondrial functions are a major pathophysiological basis of diabetic cardiomyopathy. 5′ AMP-activated protein kinase (AMPK) is involved in mitochondrial dynamics. As an activator of AMPK, this study examined the effect of metformin on cardiomyocytes treated with high glucose. Primary cardiomyocytes isolated from neonatal rat ventricles were exposed to a high glucose concentration (33 mM) to establish a model of high-glucose injury with or without metformin (2 mM) treatment. AMPK activity was inhibited or activated by CC (20 µM) or AICAR (50 µM). CCK-8 and TUNEL assays were used to assess cell viability and apoptosis, respectively. A JC-1 assay was used to measure the mitochondrial membrane potential, and MitoSOX™ staining was used to examine mitoROS. Mito-Tracker Green-stained mitochondria were visualized by confocal microscopy to assess mitochondrial fission. Furthermore, we measured the expression levels of AMPK-mediated mitochondrial dynein and apoptotic proteins by western blotting. Our results showed that AMPK activity was significantly decreased in cardiomyocytes under the high-glucose condition, which was accompanied by increased mitochondrial fragmentation and aggravated mitochondrial dysfunction. The mitochondrial membrane potential was decreased and oxidative stress was increased, leading to apoptosis. Activation of AMPK by either metformin or AICAR reversed myocardial mitochondrial dysfunction and inhibited apoptosis under high glucose. Furthermore, inhibition of AMPK activity abrogated the protective effect of metformin against high glucose–induced mitochondrial dysfunction and apoptosis in cardiomyocytes. Our study demonstrates that metformin protects cardiomyocytes from high glucose–induced mitochondrial fragmentation and apoptosis by activating AMPK.
Impact Statement
Numerous studies have confirmed that diabetic cardiomyopathy is an important cardiovascular complication in diabetic patients. There is an increasing evidence that mitochondrial dysfunction is a key event of diabetic cardiomyopathy. As an activator of AMP-activated protein kinase (AMPK), metformin has been shown to alleviate the progress of diabetic cardiomyopathy. Our data suggest that metformin protects cardiomyocytes from apoptosis. The results further demonstrate that metformin have property to overcome the high glucose–induced mitochondrial fragmentation and apoptosis by activating AMPK. Inhibition of AMPK activity abrogates the protective effect of metformin on high glucose–induced cardiomyocytes. Therefore, the protection of mitochondria by metformin is an important factor in its anti-diabetic cardiomyopathy.
Introduction
Diabetic cardiomyopathy (DCM) is a major cause of cardiovascular complications and death of diabetic patients. 1 Although hyperglycemia is a pathological feature of diabetes and exacerbates the progression of DCM, the exact mechanisms of how hyperglycemia influences pathological processes are unclear. Healthy mitochondria are vital for energy production in cardiomyocytes. However, upon damage, mitochondria produce large amounts of reactive oxygen species (ROS) and pro-apoptotic factors.2 –4 There is increasing evidence that mitochondrial dysfunction is a crucial event of DCM.5–7 Although mitochondria are associated with DCM, the underlying molecular mechanisms remain elusive.
Excessive production of ROS in cardiomyocytes due to glucotoxicity is a major reason for the development of DCM.8–11 As the center of cellular energy metabolism, mitochondria are dynamic organelles, and fusion and fission are important physiological processes. 12 An imbalance of fusion and fission leads to abnormal mitochondrial functions, which results in apoptosis. 13 Hyperglycemia affects mitochondrial biogenesis, and glucotoxicity decreases mitochondrial fusion and increases mitochondrial fission.14–16 Conversely, inhibition of mitochondrial fission reduces high glucose–induced ROS production. 17
AMPK is an important molecule in regulation of bioenergy metabolism, implying a close link between AMPK activity and mitochondrial functions. AMPK deficiency affecting various aspects of mitochondrial biology has been found in various mouse models.18,19 AMPK activity is inhibited in the diabetic state, and as an activator of AMPK, metformin alleviates DCM in diabetic OVE26 mice by upregulating autophagic activity. 20 However, the role of metformin in DCM remains unclear, particularly in regulating mitochondrial dynamics.
In this study, we found that metformin regulates fission and fusion of myocardial mitochondria by activating AMPK and inhibits glucotoxicity-induced cardiomyocyte apoptosis. This potential molecular mechanism may provide a new direction and therapeutic target for the treatment of DCM.
Materials and methods
Primary cardiomyocyte culture and treatment
The study was approved by the Experimental Animal Ethics Committee of Fujian Medical University and was performed in accordance with institutional guidelines. Primary cardiomyocytes were isolated from the one- to three-day-old neonatal ventricles of Sprague-Dawley rats by tissue digestion and differential adhesion. 21 The ventricles were cut into pieces, minced, and then digested with 0.2% collagenase II (Sigma Aldrich, USA, V900892) for 2 h on ice. Cardiomyocytes were harvested after filtration through a 200-µm mesh cell sieve and cultured at 37°C with 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco, 10100147) for differential adhesion. To induce high glucose–mediated myocardial injury, cardiomyocytes were treated with a high glucose concentration (33 mM) for 48 h and then treated with or without metformin (0, 0.5, 1.0, and 2.0 mM) to assess its cardioprotective effect. To inhibit or promote AMPK activity, cardiomyocytes were pretreated for 5 h with compound C (20 µM, Selleck Chemicals, USA, S7306) or 5-amino-4-imidazole-1-β-D-carboxamide ribofuranoside (AICAR; 500 µM, Selleck Chemicals, S1802).
Cell viability assay
Cell viability was examined by a cell counting kit-8 (CCK-8) assay (Sigma-Aldrich, 96992). Briefly, primary cardiomyocytes were seeded at 3000 cells/well in 96-well plates. After HG exposure with or without treatment with various metformin concentrations (0, 0.5, 1.0, and 2.0 mM), 10 µL CCK-8 reagent was added to each well, followed by incubation for 2 h. Absorbance was measured at 450 nm with a microplate reader (BIO-TEX, USA, ELx800).
Measurement of mitochondrial ROS
To assess mitochondrial reactive oxygen species (mitoROS) of primary cardiomyocytes, MitoSOX™ Red Mitochondrial Superoxide Indicator (Invitrogen, USA, M36008), was used. The probe rapidly and selectively targets mitochondria and is rapidly oxidized by superoxide to produce substantial fluorescence. After treatment with normal glucose or HG with or without metformin for 48 h, cardiomyocytes were incubated with 3 µM MitoSOX™ for 10 min at 37°C. Then, the cardiomyocytes were washed with prewarmed medium and analyzed using a confocal laser-scanning microscope (Zeiss, Germany, LSM800) or flow cytometer (Biosciences, USA, AccuriC6). The level of mitoROS was represented by the mean fluorescence intensity of 40–50 cells quantified by Image-Pro Plus 6.0 software.
Measurement of the mitochondrial membrane potential
Mitochondrial membrane potential (MMP) was measured by staining with JC-1 (Beyotime Biotechnology, China, C2006). Cardiomyocytes were seeded in 12-well plates and treated with normal glucose or HG with or without metformin for 48 h. Subsequently, the cardiomyocytes were incubated in the JC-1 staining solution for 30 min at 37°C. After replacing the staining solution with fresh culture medium, the cells were observed under a fluorescence microscope (Olympus, Japan, CX41). The red-to-green fluorescence intensity ratio was measured as the MMP in five random fields.
TUNEL assay
Apoptosis of primary cardiomyocytes following high glucose–induced injury was analyzed by a terminal transferase UTP nick end labeling (TUNEL) assay using an In Situ Cell Death Detection Kit (Roche Diagnostics, USA, 11684795910). Cardiomyocytes in 12-well plates were washed with phosphate-buffered saline (PBS) after treatment with normal glucose or HG with or without metformin for 48 h and then fixed for 30 min in 4% paraformaldehyde. After washing with PBS, the cells were permeabilizated with 0.3% Triton X-100 in PBS for 5 min at room temperature. Then, 50 µL TUNEL detection solution was added to the cells, followed by incubation at 37°C for 1 h. DAPI was applied to visualize nuclei, and the cells were observed under the fluorescence microscope. The number of positive cells was counted in five random fields.
Western blot analysis
Protein expression was analyzed by western blotting. Cardiomyocyte proteins were collected and frozen at −80°C for western blotting analysis. The same amount of proteins was electrophoresed using 12%–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto PVDF membranes. The membranes were then incubated with blocking buffer at room temperature (5% dry milk, 0.1% Tween 20, in TRIS-buffered saline [TBS]) for 2 h and primary antibodies at 4°C overnight and then with a corresponding Horseradish peroxidase (HRP)-conjugated secondary antibody (1:8000, Proteintech, PR30011 or PR30012) at room temperature for 2 h. Protein bands were analyzed by Quantity One Software (Bio-Rad Laboratories, USA) and normalized to the internal reference protein GAPDH (1:3000, Proteintech, 60004-1-Ig). Primary antibodies were against total AMPK (1:1000, ImmunoWay Biotechnology, YT0216), Bcl2 (1:1000, Abcam, ab196495), Bax (1:1000, Abcam, ab32503), caspase 3 (1:2000, Abcam, ab184787), total Drp1 (1:2000, Abcam, ab184247), Opa1 (1:1000, Abcam, ab119685), phospho-AMPK (Thr172) (1:1000, Cell Signaling Technology, 2535), phospho-Drp1 (Ser616) (1:1000, Cell Signaling Technology, 3455), and cleaved-caspase 3 (1:1000, Cell Signaling Technology, 9664).
Measurement of mitochondrial fission
Mitochondrial fission was assessed by measuring the mitochondrial length. 22 Mitochondrial profiles were determined by Mito-Tracker Green (Beyotime Biotechnology, C1048). Mito-Tracker Green is a mitochondrion-specific fluorescent dye for live cells. After treatments, cardiomyocytes were stained with a Mito-Tracker Green staining solution (1:10,000) at 37°C for 30 min. Subsequently, the cells were washed twice with prewarmed culture medium, and the mitochondrial length in 40–50 cells was calculated using Image-Pro Plus 6.0 software.
Statistics
Data are presented as the mean ± standard deviation (SD). At least six independent experiments were performed. Statistical analyses of group comparisons were performed by one-way analysis of variance. Statistical significance was considered at P < 0.05.
Results
Metformin improves cell viability and inhibits apoptosis of cardiomyocytes induced by high glucose
To explore adverse effects of glucotoxicity on cardiomyocytes, cardiomyocytes were treated with 33 mM glucose (HG) for 12, 24, 48, and 72 h. As shown in Figure 1(A), cardiomyocytes showed a time-dependent decrease in cell viability under the HG condition with the most significant decrease at 48 h. We next explored the protective effect of metformin on cardiomyocytes. We observed a concentration-dependent improvement in cell viability of cardiomyocytes treated with high glucose for 48 h and various concentrations of metformin (0.5, 1.0, and 2.0 mM) (Figure 1(B)). High glucose treatment for 48 h increased apoptosis of cultured cardiomyocytes, and 2 mM metformin significantly inhibited cardiomyocyte apoptosis compared with the HG group (Figure 1(C) and (D)). Western blotting of cardiomyocytes showed that expression of proapoptotic proteins Bax and C-caspase 3 was upregulated, while Bcl2 expression was downregulated after HG stimulation for 48 h (Figure 1(E) and (F)). Taken together, these results suggested that metformin attenuated glucotoxicity-induced apoptosis of cardiomyocytes.
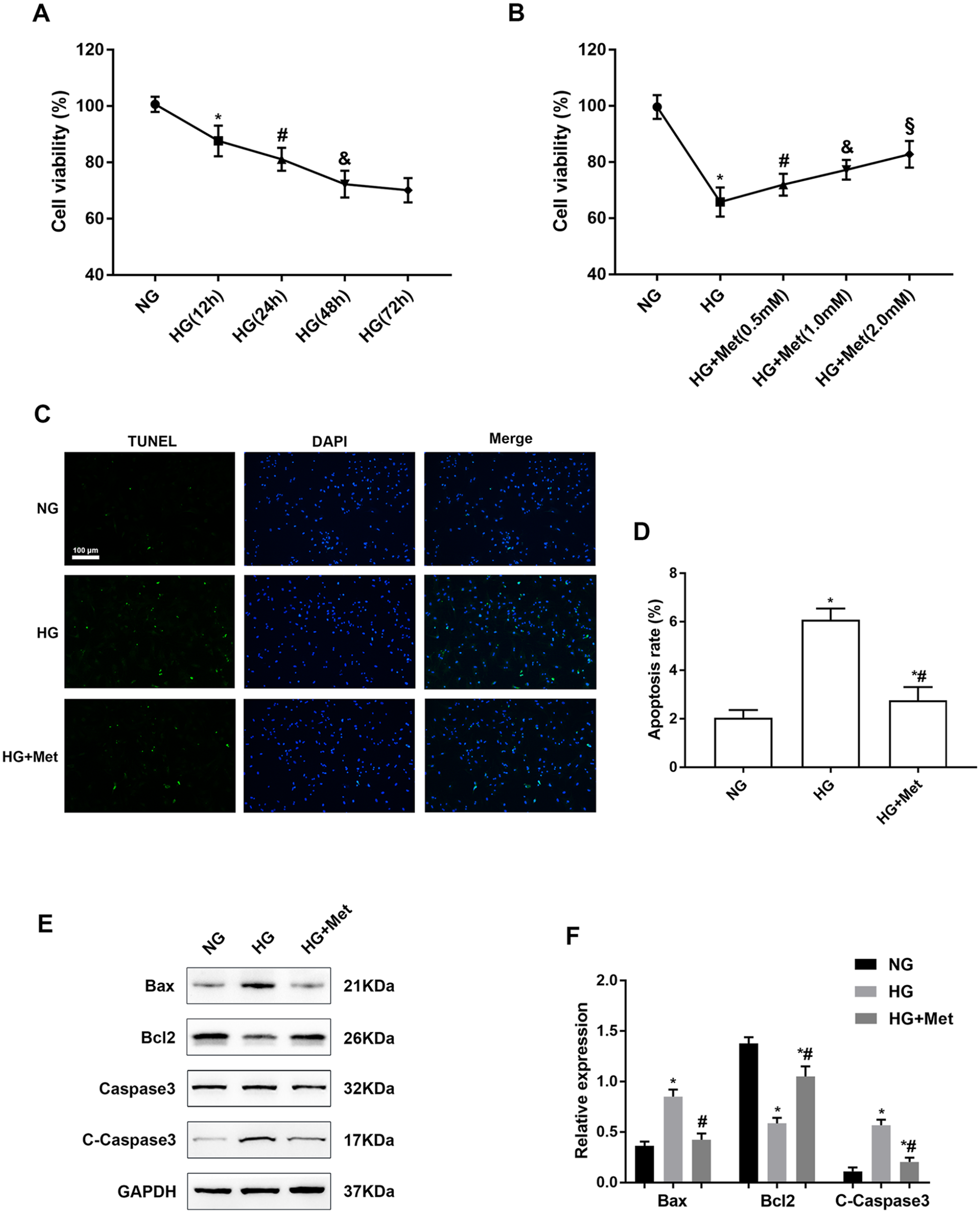
Metformin increases cell viability and inhibits apoptosis of high glucose–treated cardiomyocytes: (A) Cell viability of cardiomyocytes treated with 33 mM glucose for 12, 24, 48, and 72 h was measured by CCK-8 assays. n = 6, *P < 0.05 vs NG; #P < 0.05 vs HG (12 h); &P < 0.05 vs HG (24 h). (B) Cardiomyocytes were treated with various metformin concentrations (0.5, 1.0, and 2.0 mM) and HG for 48 h, and then cell viability was measured by CCK-8 assays. n = 6, *P < 0.05 vs NG; #P < 0.05 vs HG; &P < 0.05 vs HG + Met (0.5 mM); §P < 0.05 vs HG + Met (1.0 mM). (C) and (D) Apoptotic cells were detected by TUNEL staining. The apoptotic rate is expressed as the ratio of TUNEL-positive cells to total cells in five random fields. (E) and (F) Western blot analysis of Bax, Bcl2, caspase 3, and C-caspase 3 expression in cardiomyocytes treated with NG or HG with or without 2 mM metformin for 48 h. Grayscale ratios of Bax/GAPDH, Bcl2/GAPDH, and C-caspase 3/caspase 3 indicated the expression levels of apoptotic proteins. n = 6, *P < 0.05 vs NG; #P < 0.05 vs HG.
Metformin attenuates high glucose–induced mitochondrial dysfunction and fission in cardiomyocytes
Next, we evaluated the effect of metformin on mitochondrial functions and fission in cardiomyocytes treated with high glucose. Immunofluorescence was employed to analyze mitochondrial morphology using Mito-Tracker Green. As shown in Figure 2(A) and (B), mitochondria in the NG group were highly interconnected to form a network structure. Interestingly, mitochondria became shorter in response to high glucose stress, and this alteration was attenuated by metformin treatment. Mitochondrial fission was quantified by measuring the mean mitochondrial length. Compared with the NG group, mitochondrial length was significantly reduced after high glucose treatment, and metformin treatment prevented high glucose–induced mitochondrial fission. Subsequently, to assess the protective effect of metformin on cardiomyocyte mitochondria, the MMP was measured by JC-1 staining, and mitoROS were assessed by MitoSOX™ staining. As shown in Figure 2(C) and (D), high glucose decreased the MMP in cardiomyocytes, and metformin increased the MMP in cardiomyocytes. We also found that mitoROS in cardiomyocytes after high glucose stimulation were significantly increased compared with the NG group, and metformin inhibited the high glucose–induced increase in mitoROS (Figure 2(E) and (F)). These findings demonstrated that metformin protected cardiomyocytes from high glucose–induced mitochondrial dysfunction.
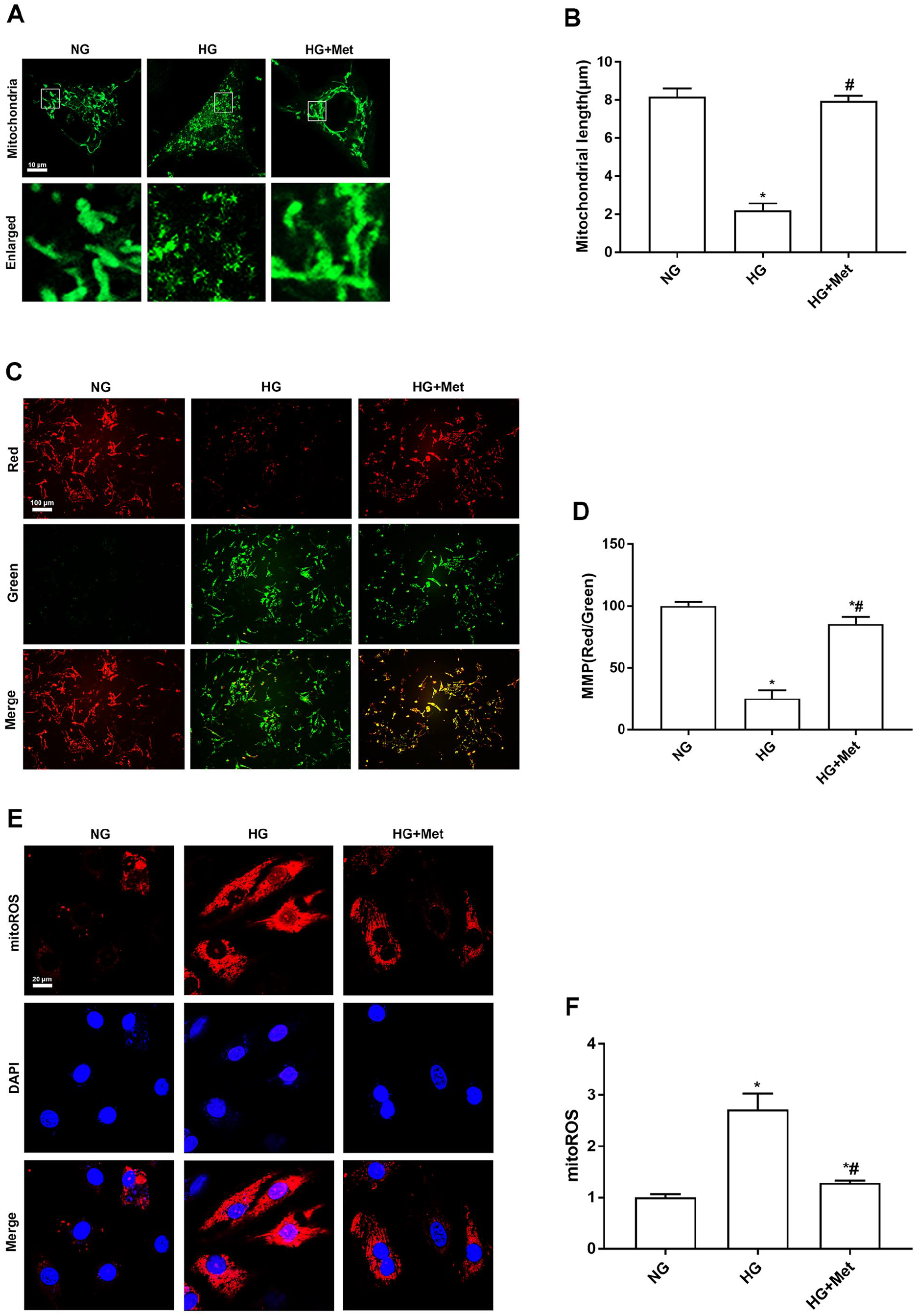
Metformin attenuates mitochondrial dysfunction and fission in high glucose–treated cardiomyocytes. (A) and (B) After cardiomyocytes were treated with NG or HG with or without 2 mM metformin for 48 h, mitochondrial fission was observed by Mito-Tracker Green staining using confocal microscopy. The mean mitochondrial length was used to quantify mitochondrial fission. (C) and (D) MMP in cardiomyocytes was determined by JC-1 staining represented as the ratio of red-to-green fluorescence. (E) and (F) MitoSOX™ staining was used to analyze the level of mitoROS determined by the relative fluorescence intensity. n = 6, *P < 0.05 vs NG; #P < 0.05 vs HG.
Metformin upregulates AMPK activity and affects the expression of mitochondrial dynein in high glucose–treated cardiomyocytes
To evaluate the effect of metformin on AMPK activity and mitochondrial dynein expression, we conducted western blotting. High glucose decreased AMPK activity in cardiomyocytes, which was reversed by metformin treatment. In addition, compared with the NG group, the phosphorylation level of Ser616 in mitochondrial fission protein Drp1 was increased in HG-treated cardiomyocytes, and expression of mitochondrial fusion protein Opa1 was decreased. Metformin treatment upregulated the expression of Opa1 and downregulated the phosphorylation level of Drp1. These results suggested that metformin promoted cardiomyocyte fusion in high glucose–treated cardiomyocytes (Figure 3(A) to (D)).
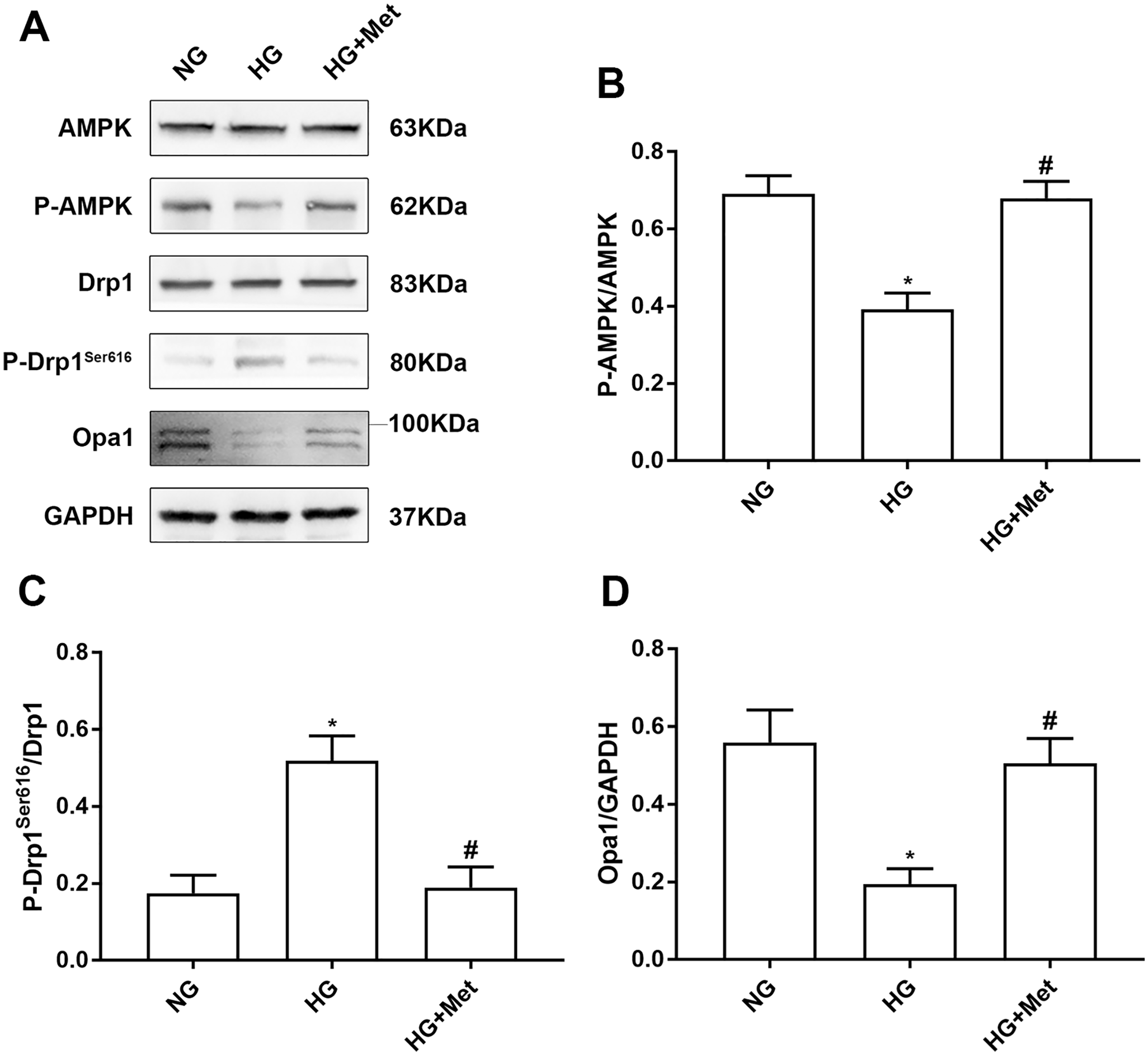
Metformin upregulates AMPK activity and affects the expression of mitochondrial dynein in high glucose–treated cardiomyocytes. (A) to (D) Western blotting was used to measure the level of related proteins in cardiomyocytes treated with NG or HG with or without metformin for 48 h. n = 6, *P < 0.05 vs NG; #P < 0.05 vs HG.
Inhibition of AMPK abolishes the effect of metformin preventing mitochondrial fission and mitochondrial dysfunction in high glucose–treated cardiomyocytes
We next determined whether metformin attenuated high glucose–induced mitochondrial fission and mitochondrial dysfunction in cardiomyocytes via AMPK activation using the AMPK inhibitor CC and AMPK activator AICAR. As shown in Figure 4(A) to (D), western blotting demonstrated that activation of AMPK by AICAR alleviated the upregulated phosphorylation of Drp1 and downregulated expression of Opa1 in cardiomyocytes under high glucose. Moreover, CC abolished metformin-downregulated Drp1 phosphorylation and upregulated Opa1 expression by inhibiting AMPK activity. Subsequently, confocal laser microscopy showed that AMPK activation alleviated mitochondrial fission compared with the HG group and inhibition of AMPK activity abolished the fusion effect of metformin on mitochondria (Figure 4(E) and (F)). Furthermore, we assessed the protective effect of AMPK activation on MMP by JC-1 staining. As shown in Figure 4(G) and (H), inhibition of AMPK activity abolished upregulation of the MMP by metformin, and activation of AMPK alleviated the downregulated MMP by high glucose. Next, the mitoROS level was determined by flow cytometric analysis of the relative fluorescence intensity. AICAR treatment attenuated the high glucose–induced mitoROS level by activating AMPK, and CC treatment abolished the effect of metformin on reducing mitoROS (Figure 4(I) and (J)). These results confirmed that metformin attenuated high glucose–induced mitochondrial fission and mitochondrial dysfunction in cardiomyocytes via AMPK activation.
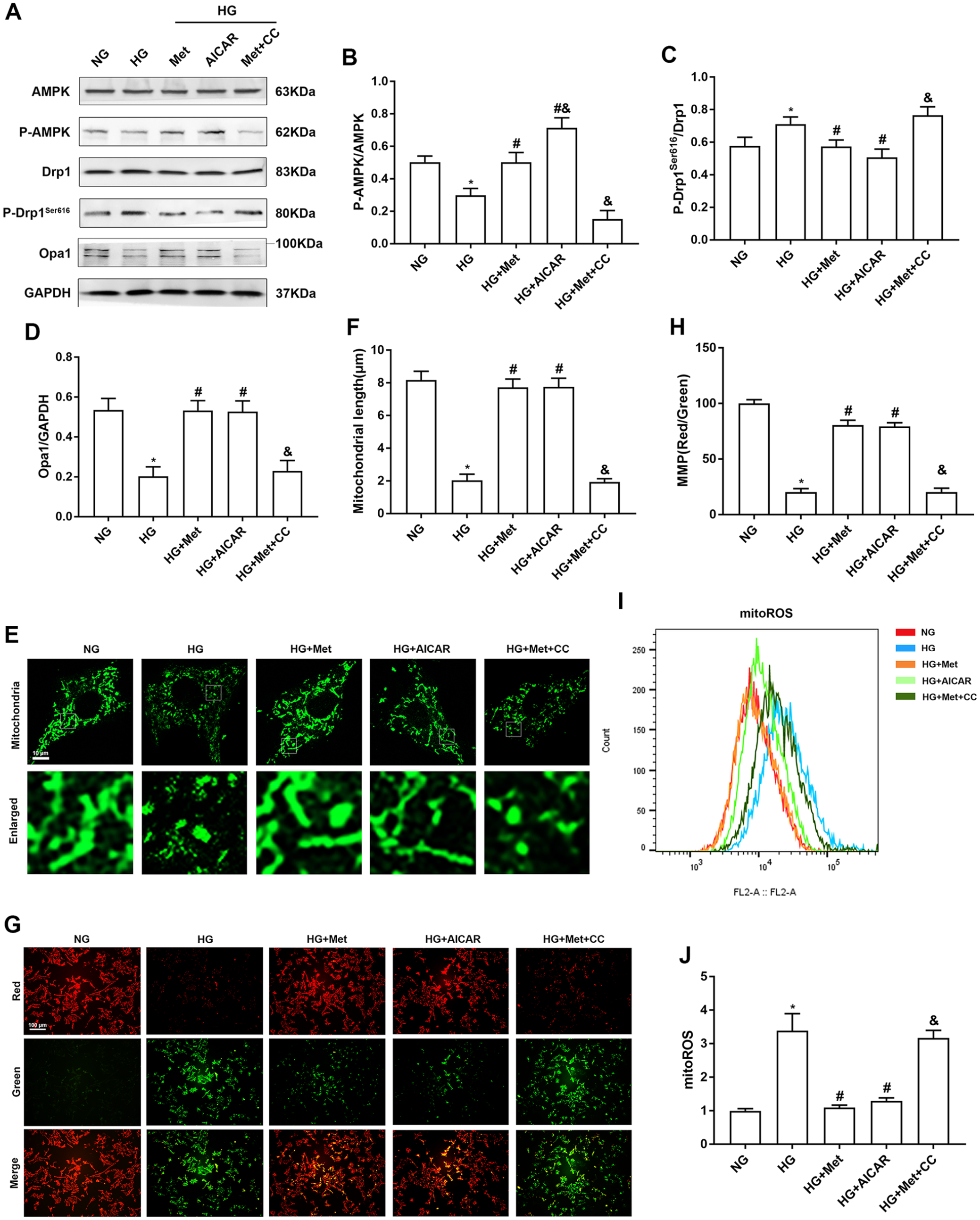
Inhibition of AMPK abolishes metformin preventing mitochondrial fission and dysfunction in high glucose–treated cardiomyocytes. Cardiomyocytes were pretreated with CC (20 µM) or AICAR (500 µM) for 5 h, followed by treatment with NG or HG with or without metformin for 48 h. (A) to (D) Western blot analysis of AMPK, p-AMPK, Drp1, P-Drp1Ser616, and Opa1 expression in cardiomyocytes. (E) and (F) Mitochondrial fission was observed by Mito-Tracker Green staining using confocal microscopy. The mean mitochondrial length was used to quantify mitochondrial fission. (G) and (H) MMP was determined by JC-1 staining represented as the ratio of red-to-green fluorescence. (I) and (J) MitoSOX™ staining was used to analyze the level of mitoROS determined by flow cytometric analysis with at least 10,000 cells per group. n = 6, *P < 0.05 vs NG; #P < 0.05 vs HG; &P < 0.05 vs HG + Met.
Inhibition of AMPK abrogates the effect of metformin attenuating apoptosis in high glucose–treated cardiomyocytes
To verify whether metformin attenuating high glucose–induced cardiomyocyte apoptosis was dependent on AMPK activation, we used AMPK inhibitor CC and AMPK activator AICAR. Western blotting showed that expression of Bax and C-caspase 3 was decreased and Bcl2 expression was increased by AICAR treatment compared with the HG group. CC treatment abolished the anti-apoptotic effect of metformin on high glucose–treated cardiomyocytes, and expression of Bax and C-caspase 3 was increased and Bcl2 expression was decreased (Figure 5(A) to (D)). TUNEL assays showed that AICAR treatment alleviated apoptosis of high glucose–treated cardiomyocytes and exerted a similar effect to metformin. CC attenuated the inhibitory effect of metformin on cardiomyocyte apoptosis (Figure 5(E) and (F)).
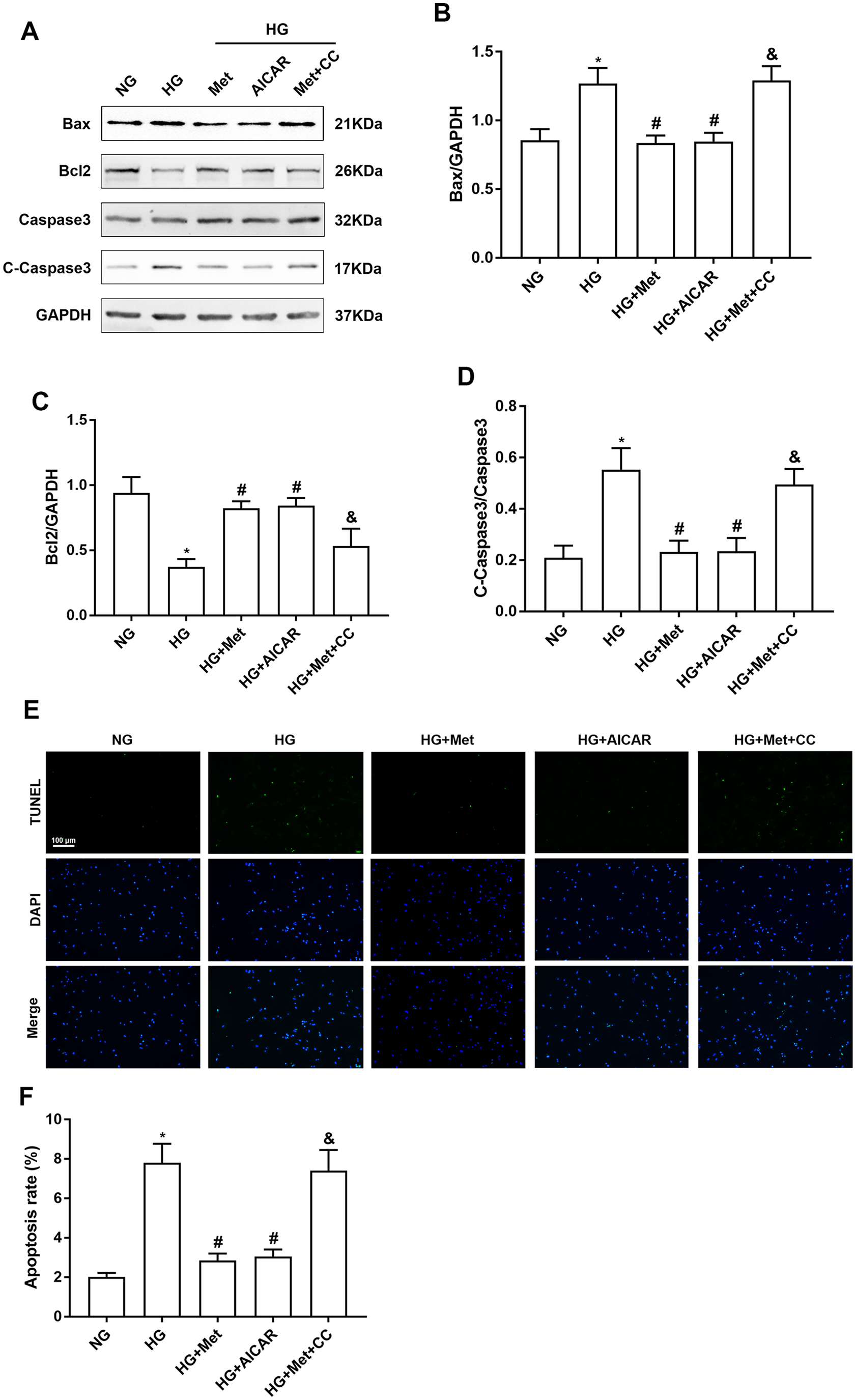
Inhibition of AMPK abrogates metformin attenuating apoptosis in high glucose–treated cardiomyocytes. Cardiomyocytes were pretreated with CC (20 µM) or AICAR (500 µM) for 5 h, followed by treatment with NG or HG with or without metformin for 48 h. (A) to (D) Western blot analysis of Bax, Bcl2, caspase 3, and C-caspase 3 expression in cardiomyocytes. (E) and (F) TUNEL assay to detect apoptosis of cardiomyocytes. n = 6, *P < 0.05 vs NG; #P < 0.05 vs HG; &P < 0.05 vs HG + Met.
Discussion
DCM is a major cardiac complication and cause of death of patients with diabetes, and its pathogenesis is very complex. With the deepening of research, increasingly more researchers believe that mitochondrial dysfunction is a major factor3,4,23 In this study, we found that metformin improved mitochondrial fission and fusion as well as mitochondrial functions by activating AMPK, thereby protecting cardiomyocytes from high glucose–induced apoptosis in vitro. The positive effect of metformin on mitochondria might play an important role in hyperglycemia-induced cardiomyopathy. The use of primary cardiomyocytes in vitro in this study makes the results more convincing. This study is innovative by exploring the protective effect of metformin on high glucose–treated cardiomyocytes from the perspective of mitochondrial dynamics.
In addition to its hypoglycemic effect, metformin also exerts anti-aging, anti-tumor, and cardioprotective effects.24–26 Particularly in protection against DCM, metformin reduces myocardial cell death, corrects metabolic disorders, and reduces inflammation and oxidative stress, thereby improving the cardiac structure and functions.27–29 In this study, high glucose induced apoptosis of cardiomyocytes, which was reduced by metformin treatment. In addition, the anti-apoptotic effect of metformin might be maintained through protection of myocardial mitochondria.
Mitochondria generate adenosine triphosphate (ATP) in cardiomyocytes to maintain metabolism and contraction. Therefore, mitochondrial dysfunction is closely related to various cardiovascular diseases including DCM, ischemic reperfusion myocardial damage, and heart failure.30–32 Mitochondrial dynamics play a very important role in maintaining the mitochondrial bioenergetic function,33,34 and increased fission or decreased fusion leads to mitochondrial fragmentation and subsequent mitochondrial dysfunction. Excessive mitochondrial fission is a major factor in the diabetic heart, and regulation of mitochondrial dynamics may be a potential therapeutic strategy for DCM. 16 In this study, after exposure of cardiomyocytes to high glucose, we observed a decrease in mitochondrial fusion and an increase in mitochondrial fission. Moreover, the MMP was decreased and mitoROS were increased. However, after metformin treatment, mitochondrial fission was reduced and mitochondrial fusion was increased. The MMP was also increased, while mitoROS were decreased. These results suggest that metformin improves myocardial mitochondrial dynamics and functions.
As a member of the serine/threonine (Ser/Thr) kinase family, AMPK is a crucial regulator of cardiac energy homeostasis and plays an important role in reducing apoptosis of cardiomyocyte.35,36 In vitro and in vivo studies have found downregulation of AMPK in post-infarct cardiac injury, and correcting the AMPK-Drp1 axis and inhibiting mitochondrial fission might be an effective approach to delay the development of post-infarct cardiac injury. 37 Furthermore, AMPK activation alleviates myocardial ischemia-reperfusion injury by promoting Opa1-related mitochondrial fusion. 38 In our study, exposure of cardiomyocytes to a high glucose environment decreased AMPK activity, and AMPK activity was significantly improved by metformin treatment that also suppressed phosphorylation of Drp1 and promoted expression of Opa1. After inhibition of AMPK activity by compound C, the effects of metformin in improving mitochondrial function and inhibiting apoptosis were abolished. Interestingly, the AMPK activator AICAR had a similar protective effect to metformin on cardiomyocytes, suggesting that the anti-apoptotic effect of metformin was dependent on AMPK activation.
Although our study found that metformin improved cardiac functions by regulating mitochondrial dynamics as shown in Figure 6, there are several limitations. This study was limited to cell experiments and lacked in vivo experiments to support the results. We also mainly focused on mitochondrial dynamics but did not observe other aspects of mitochondria, such as mitophagy. In addition, the specific mechanism by which metformin regulates DCM remains unclear and requires further research.
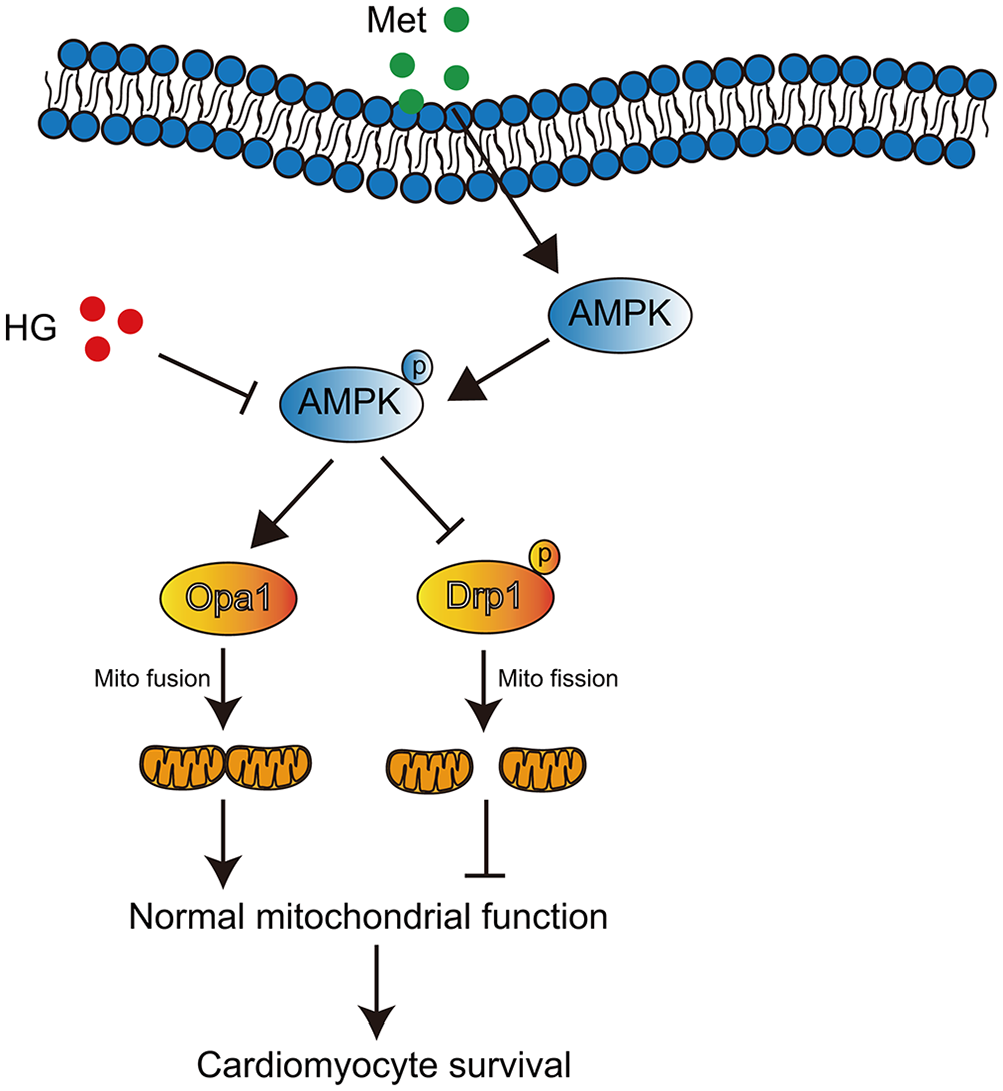
Schematic representation of a possible molecular model for metformin in inhibiting cardiomyocyte apoptosis induced by high glucose.
Footnotes
Authors’ Contributions
YW designed and conducted the study, analyzed the data, and wrote the manuscript.
Declaration Of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Startup Fund for scientific research, Fujian Medical University (grant no. 2019QH2026); and the Funding for Top Hospital and Specialty Excellence of Fujian Province.