
Abstract
High-throughput genome-wide sequencing has revealed high genomic variability of HPV16 in different geographic regions which is the most predominant genotype in human papillomavirus (HPV)–associated malignancies. Analysis of the HPV16 by whole-genome sequence (WGS) is an advanced method for the identification of mutations in the genome. There is limited information about HPV16 diversity in Pakistan, especially at the genomic level. Till now, WGS for HPV16 has not been previously reported in Pakistan. The current study has sequenced three HPV16 viral genomes, from two cervical and one oral cavity positive sample of women presented with general gynecological problems without any evidence of precancerous or cancerous lesions using an ion ampliseq customized panel. Sequencing analysis detected 38 variations, including single-nucleotide polymorphisms (SNPs) and two Indels, across three samples with the highest number of SNPs present in E1, E2, and L2, respectively. A total of 20 non-synonymous and 11 synonymous mutations with amino acid substitutions (T1421C, G1515A, T2223C, T1389C, G1483A, and T2191C) were identified. The phylogenetic analysis revealed the genomes of HPV16 are closely associated with those reported from Thailand and the United States. These are the first HPV16 WGS from Pakistan. However, more research is needed with a large sample size from diversified areas to assess the carcinogenic consequences and impact of HPV vaccinations.
Keywords
Impact statement
This study identifies novel variants in HPV16 genome from Pakistani subjects using whole-genome sequence (WGS) and deciphering its role in disease pathology. The phylogenetic analysis revealed the evolutionary history of virus transmission directing to deliver basic data for future studies on their distinct epidemiology and evolution
Introduction
Human papillomavirus (HPV) infections are the most common sexually transmitted disease in the world characterized by cervical, oropharyngeal, penile, vulvo-vaginal, and anal cancers. 1 In 2020, cervical cancer (CC) was the fourth most diagnosed cancer in the world and the third most common tumor among females in Pakistan where HPV plays a pivotal role in its development. 2 Likewise, the incidence of oral cancer is more frequent in South-Central Asia (India and Pakistan). 2 The current status and data of “high-risk” HPV (HR-HPV) prevalence in normal oral cavity and cervical among the Pakistani population are, however, limited.3–6
The HPV is a group of more than 200 related viruses differentiated by genetic diversity. 7 These are further classified as “low-risk” HPV (LR-HPV) and HR-HPV types. 8 Among 13 different types of HPV genotypes, HPV-16 is most commonly detected in cervical cancer accounting for nearly 60% of invasive cervical cancers worldwide. HPV16 is well documented as HR-HPV because of its raised carcinogenic properties and is known to be observed in more than 70% of low-grade CIN but persistent HPV infection might cause cellular changes which lead to high-grade cervical intraepithelial neoplasia (CIN) and invasive cervical cancer. 9 Furthermore, HPV16 is also known to be associated with head and neck squamous cell carcinomas. 10
The double-stranded (dsDNA) HPV viral genome replicates as an extrachromosomal plasmid in the nucleus of infected keratinocytes. 8 It varies in size among different HPV genotypes, but they are typically 8 kb in length, with a diameter of 52–55 nm and an icosahedral capsid made of 72 capsomeres. 8 The viral genome is divided into three sections. The first section is called the early (E) region (E1–E8 genes) which encodes proteins required for viral replication, transcription, and translation, and proteins required for host cell genomic instability by interfering with normal cell cycle regulation. 11 The second division is the late region (L1 and L2 genes) that codes for viral structural proteins of capsids and the formation of new virion particles or genome packing in viral capsids. The last region is the non-coding long control region (LCR).11,12
HPV novel types are assigned by a unique number based on the L1 sequence by the International HPV Reference Center. Variation in DNA sequences in the L1 region is observed in 10% of each HPV genotype.7,13 Based on sequence comparison of the L1 gene (the most conserved region of the HPV genome), novel HPV genotypes are assigned their specific taxonomy. If the L1 Open Reading Frame (ORF) region sequence differs by 10% or more, it is considered a novel papillomavirus. 8 However, if the nucleotide differences among the HPV sequences are less than 10%, they are believed to be the same HPV types but can be categorized into lineages (1–10%) and sublineages (0.5–1%). 14 Initially, evolutionary intra-typic studies related to HPV variants were confined to E6, E7, and LCR regions due to traditional screening methodologies. 15 However, with the advent of next-generation sequencing (NGS), HPV variant lineages and sublineages are examined at the whole-genome level. 14
Based on whole-genome sequence (WGS) HPV, 16 variants have been categorized into four major lineages A to D and their sublineages are A1–3 (European), A4 (Asian), B (African 1), C (African 2), D with Asian American (AA) and North American (NA).14,16 It is also essential to emphasize that multiple HPV variants of the distinct phylogenetic lines are dispersed throughout the world unequally. 17
In our previous study, more than 50% HR-HPV prevalence (n = 51) was observed in tested cervical samples collected from women of different cities of Punjab reporting to Lady Willingdon Hospital of Lahore, Pakistan. The prevalence of genotypes was found as HPV 16 (18%), HPV 18 (6%), and HPV 45 (1%). 18 In this work, three well-characterized HPV-16-positive cervical and oral samples from females with general gynecological issues and no signs of precancerous or cancerous lesions were analyzed using the WGS method. In the investigated samples, the HPV-16 lineages and sublineages were also molecularly defined by this WGS analysis. In addition, evolutionary phylogenetic analysis was discussed, with the aim of providing fundamental information for next studies on their unique epidemiology and evolution.
Materials and methods
Participant information
This cross-section study consisted of 150 females who were sexually active, married, and visited with gynecological problems (lower abdominal pain, abnormal vaginal discharge, post coital bleeding, and heavy menstrual bleeding) at Lady Willingdon Hospital, during August 2018–September 2019. Females were informed about the study and that their participation was voluntary. Females with chronic illnesses, autoimmune diseases, a history of cervical or oral cancer, a history of chemotherapy and radiotherapy, a preneoplastic appearance of the cervical or oral mucosa, menstruation at the time of sampling, and pregnant females were excluded.
Sample collection
Clinical examination of the cervical and oral mucosa was done before sample collection. Two samples, one from each anatomical site (cervix and oral cavity), were collected from each study participant. The cervicovaginal swabs were collected from all females with a cervical swab collection kit (Puritan UniTranz-RTTM) and were placed in a labeled 5 mL vial containing a universal viral transport medium (VTM). The 2 mL of the whole saliva was collected in labeled sterile falcon tubes by means of the resting drooling method (minimal oral movements). 19 The samples were immediately placed in a transport box containing ice packs to avoid degradation of salivary proteins and later saved at –20°C until further processing.
DNA preparation
Viral DNA was extracted from 200 μL of samples using Viral Nucleic Acid Extraction Kit “Vivantis GF-1” (Vivantis technologies, Malaysia). DNA quantification was performed by Nano-Drop (Nano-Drop Technologies, Wilmington, DE, USA) and Qubit 3.0 fluorometer Assay (Invitrogen, CA, USA).
HPV typing
The HR-HPV typing (to determine HPV 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, and 68) was completed from both samples by 14 Real-TM Quant Kit (Sacace Biotechnologies, Scalabrine, Italy) according to the manufacturer’s protocols.
Whole-genome sequencing
Whole-genome sequencing from four cervical and two whole saliva samples (oral cavity) was performed using the Ion Torrent platform (ThermoFisher Scientific). The library was created using Ion AmpliSeq customized panel for HPV16, designed using the Ion AmpliSeq Designer (https://www.ampliseq.com/,LifeTechnologies) using Ion AmpliSeq™ Library Kit 2.0 (ThermoFisher Scientific) and Ion Xpress™ Barcode Adapter 1–96 Kit (ThermoFisher Scientific) according to the manufacturer’s instructions. Equimolar amplicon libraries of six samples were pooled for automated template preparation and chip loading on Ion Chef System and sequenced on an Ion GeneStudio™ S5 System using Ion 30™ Chip (ThermoFisher Scientific).
De novo assembly of HPV genomes
After sequencing, the quality of the raw reads was assessed using FastQC (Supplemental file: 1). Low-quality reads were identified and trimmed using FastQC. 20 High-quality reads were mapped against a reference genome of HPV type 16 using Burrows–Wheeler Alignment (BWA). 21 HPV-paired clean reads were then assembled using Velvet 1.2.10 22 assembler at default parameters.
Mutation analysis
After the sequence mapping, the DNA variant regions were piled up with Torrent Variant Caller plug-in software set to run at high stringency. The depth of coverage information was obtained by Torrent Coverage Analysis plug-in software (ver. 5.0.4) with custom AmpliSeq™ panel information. Individual mutation coordinates were manually changed based on the first nucleotide in a distinct genome rather than a universal reference genome. The single-nucleotide variants (SNVs) that include insertions, deletions, and substitutions were all considered as variations.
The nucleotide diversity (π) for each single-nucleotide polymorphism (SNP) was calculated by dividing the number of SNPs identified in each region by the size of that region (E1, E2, E4, E5, E6, E7, L1, L2) and then taking an average across the total number of sites to get an approximate nucleotide diversity per bp using the formula developed by Begun et al. 23
PCR amplification and Sanger sequencing
To authenticate the HPV variant, we designed primers (MY9+: 5′-CGT CCA AAA GGA AAC TGA GC-3′; MY11+: 5′-GCA CAG GGA CAT AAC AAT GG-3′) to amplify the L1 region by PCR. PCR products were sequenced bidirectionally by Sanger sequencing on Seq-Studio Genetic Analyzer (Applied Biosystems). All sequences were blasted by the NCBI human mega blast database alignment tool.
Phylogenetic analysis of HPV 16
Phylogenetic analysis was performed using IQ-Tree. 24 The tool uses a rapid stochastic algorithm to infer phylogenetic trees by maximum likelihood. The percentage of trees in which the linked taxa clustered together is presented next to the branches. The phylogenetic tree was also reconstructed using the PhyML method included in the Geneious 8.1 software. The C50, C9, and O122 (GTR + R2) trees were all studied together.
Statistical analysis
Statistical analysis was performed by IBM SPSS version 22 (IBM Corp., Armonk, NY, USA). The sociodemographic characteristics and sexual attitudes of the participants were noted on a designed questionnaire. Chi-square or Fisher’s exact test was employed to determine the association of HR-HPV infection with the above-stated characteristics. A P value of ⩽0.05 was considered statistically significant.
Results
HR-HPV genotypes
HPV16 WGS were obtained from previously collected cervicovaginal swabs from a study of HR-HPV distribution in rural Lahore, Punjab, Pakistan. 18 HPV16 characterized the largest proportion of HPV infections in that cohort; 5% of whole saliva samples testing positive for high-risk oral human papiilomavirus (HR-OHPV) with three samples had a single HPV16 infection, while n = 2 (2%) had HR-OHPV co-infections with each HPV16 & 18 and HPV16 & HPV 51, respectively. Of these, two and 32 co-infection samples from both sites were excluded to evaluate for HPV16 variants associations with cytological testing.
De novo assembly
Of all samples positive for HPV16 from both sites, whole HPV16 genomes were successfully sequenced from three samples. Three of the six samples, that is, two from the cervical and one from the oral cavity effectively retrieved the HPV16 WGS, whereas the remaining three samples from the cervical and oral cavity had very low or no sequence reads and were thus excluded from the study. The FastQC report of all samples is enclosed in a supplemental file: 1. Two of the viral genomes from the cervical were assembled to full length (7.9 kb), while the one from the whole saliva was built as a near full-length genome (7.1 kb), which had all genes but lacked some non-coding sections at the beginning and end of the circular genomes based on the reference genome of HPV16 (NC 001526) (Table 1). The sequencing data obtained in this study are deposited in GenBank (GenBank Accession Numbers: MZ447799, MZ447800, and MZ447801).
Sizes of the three assembled genomes and the genes.
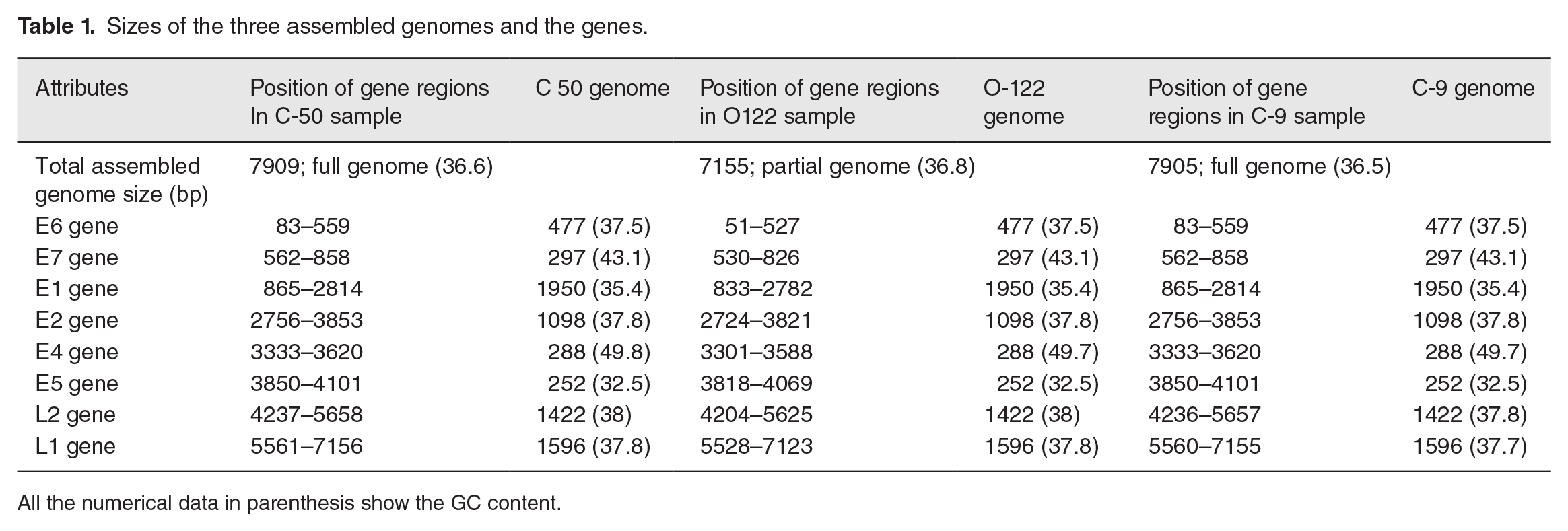
All the numerical data in parenthesis show the GC content.
The genome sizes of HPV16 sequences from cervical samples C50-PK and C9-PK were 7905 and 7909 bp (Figures 1 and 2). The genome size of HPV16 sequences from the whole saliva sample (O122-PK), was 7155 bp in size (Figure 3). The HPV16 genome has early and late regions that correspond to the position of the genes within the genome. All genes are unidirectional, except for the E4 gene, which is entirely contained within the E2 gene. The HPV16 oral genome contained a full coding part; the notable difference in size with the reference genome was due to the non-coding region, which was not detected in the HPV16 genome from the oral cavity (Figure 3).
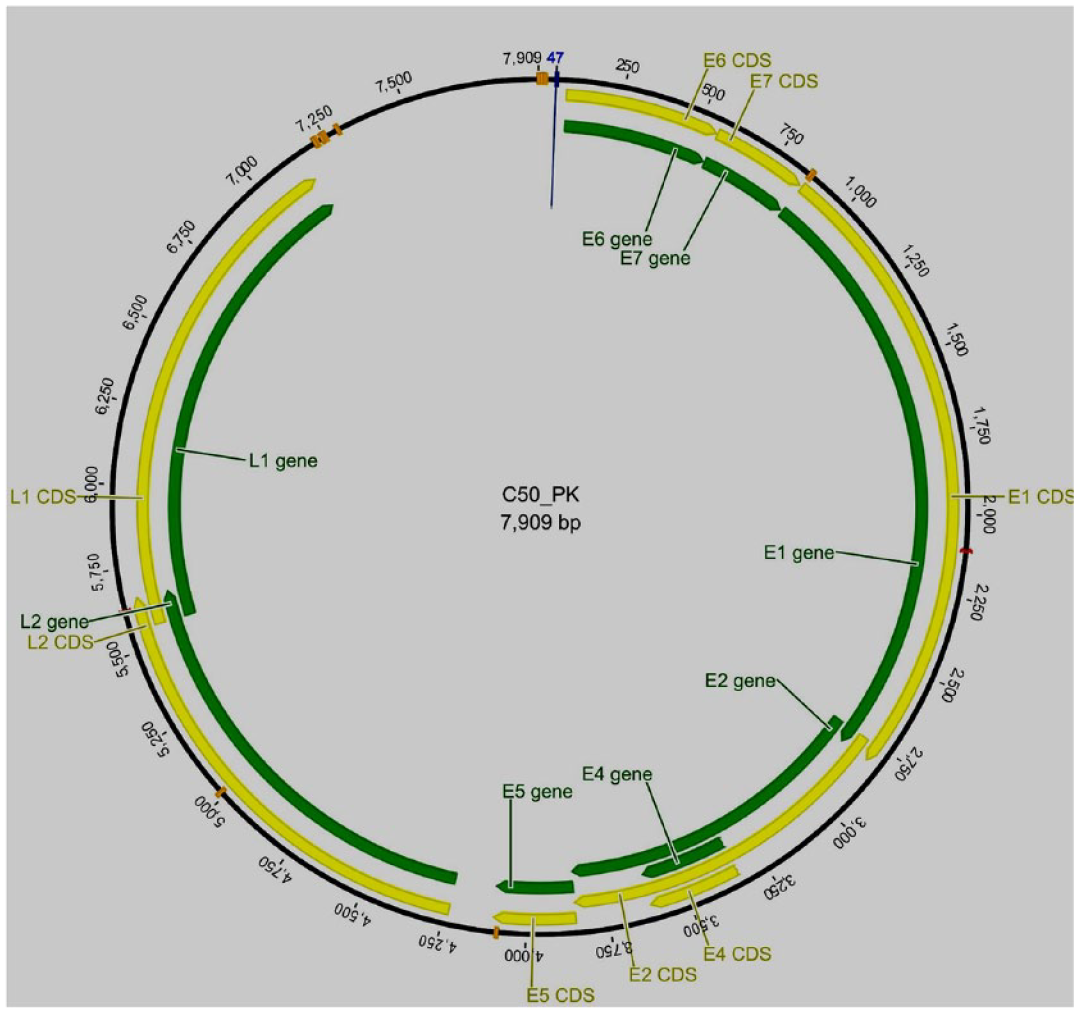
Idiograms of the cervical sample (C50_PK, HPV16) whole assembled genome sequence. The yellow lines represent coding regions or CDs, and the green bar represents the gene area.
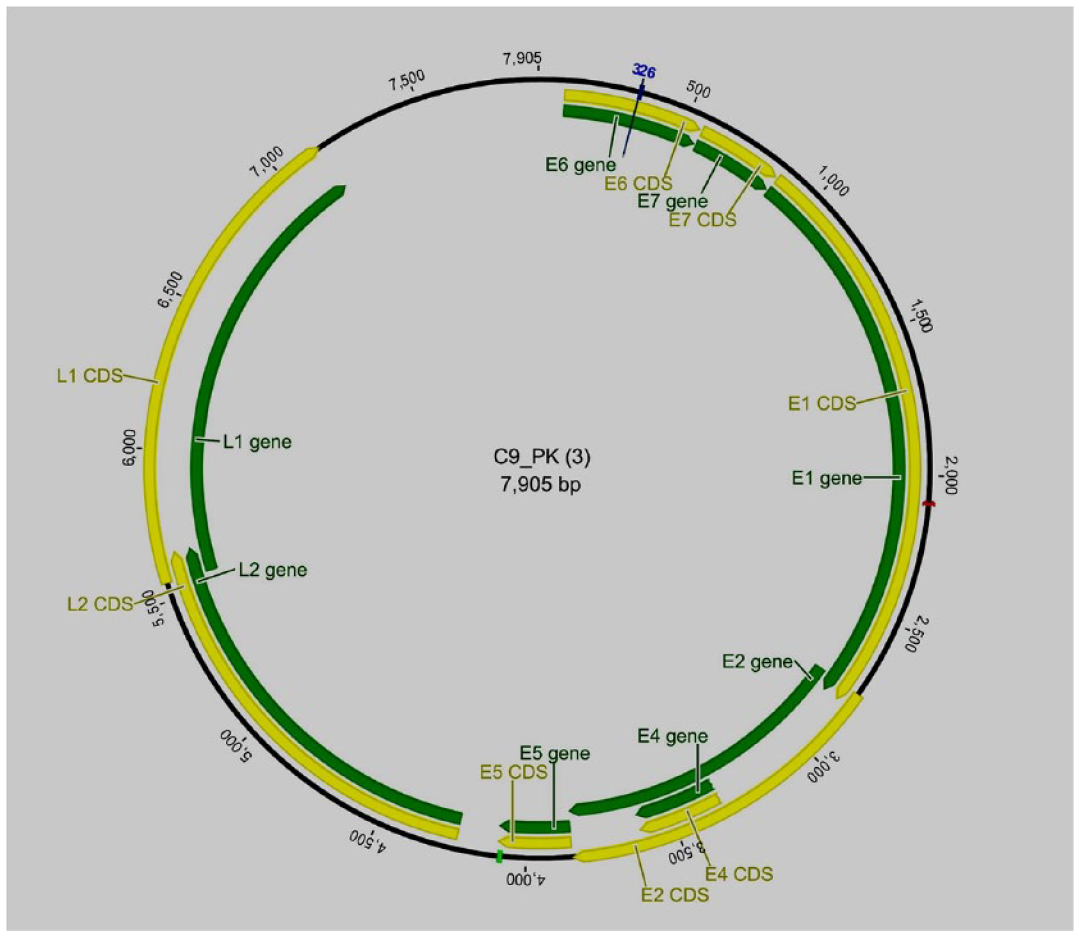
Idiograms of the cervical sample (C9_PK, HPV16) whole assembled genome sequence. The yellow lines represent coding regions or CDs, and the green bar represents the gene area.
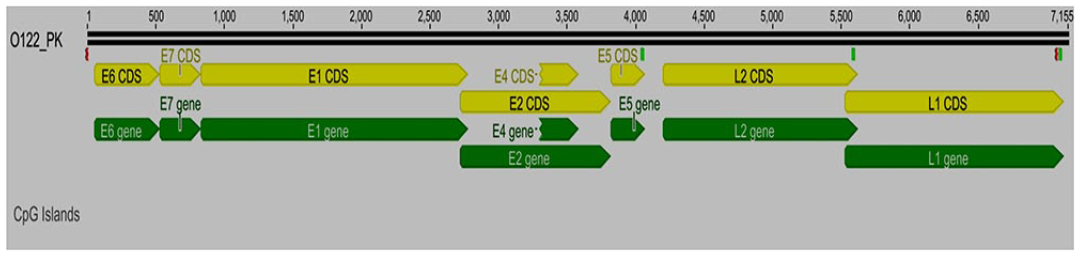
Idiograms of the oral sample (O122_PK, HPV16) whole assembled genome sequence. The yellow lines represent coding regions or CDs, and the green bar represents the gene area.
Mutation and phylogenetic analysis
Compared with the HPV16 prototype references (NC_001526), 38 variations in the three samples were observed, including 36 SNPs and two indels (insertion/deletion). E1 had the highest number of SNPs, followed by E2 and L2, while E5 and L1 had the lowest number of SNPs when compared to other HPV 16 genome regions. The nucleotide diversity study of three samples revealed that the E5 gene had the highest genetic diversity (0.0066) compared to other HPV genes, followed by the E2 (0.0018), E1 (0.0015), L2 (0.0014), and L1 (0.0010) genes. This demonstrates that changes are not evenly distributed throughout the HPV16 genome and, in particular, target HPV genome sub-regions. The majority of SNPs (31 SNPs) were in the coding region, whereas five SNPs were in the non-coding region. In all three HPV16 samples, there were 20 non-synonymous and 11 synonymous mutations. The E5 and L2 genes have the most non-synonymous mutations (n = 5), followed by the L1 gene (n = 4), and the E1 and E2 genes (n = 3). In the E4, E6, and E7 areas, no non-synonymous mutations were found. Except for one heterozygous mutation observed in HPV16 from an oral sample, all other mutations were homozygous.
The gene variants of the C9-PK sample are described in Table 2. A high number of variations were found in the coding (nine SNPs) region. The maximum number of these nucleotide sequence variations were observed in the E1, followed by the E2 and E5 regions. In contrast to synonymous mutations, the majority of the mutations were non-synonymous where T > C (4/11; 36.3 %) SNP was the most prevalent variant, followed by G > A and A > G (2/11; 18.1%), respectively. Interestingly, the C9-PK sample was found to have five unique SNPs that had not previously been reported.
Mutations detected in the HPV16 samples from cervical and oral cavity.
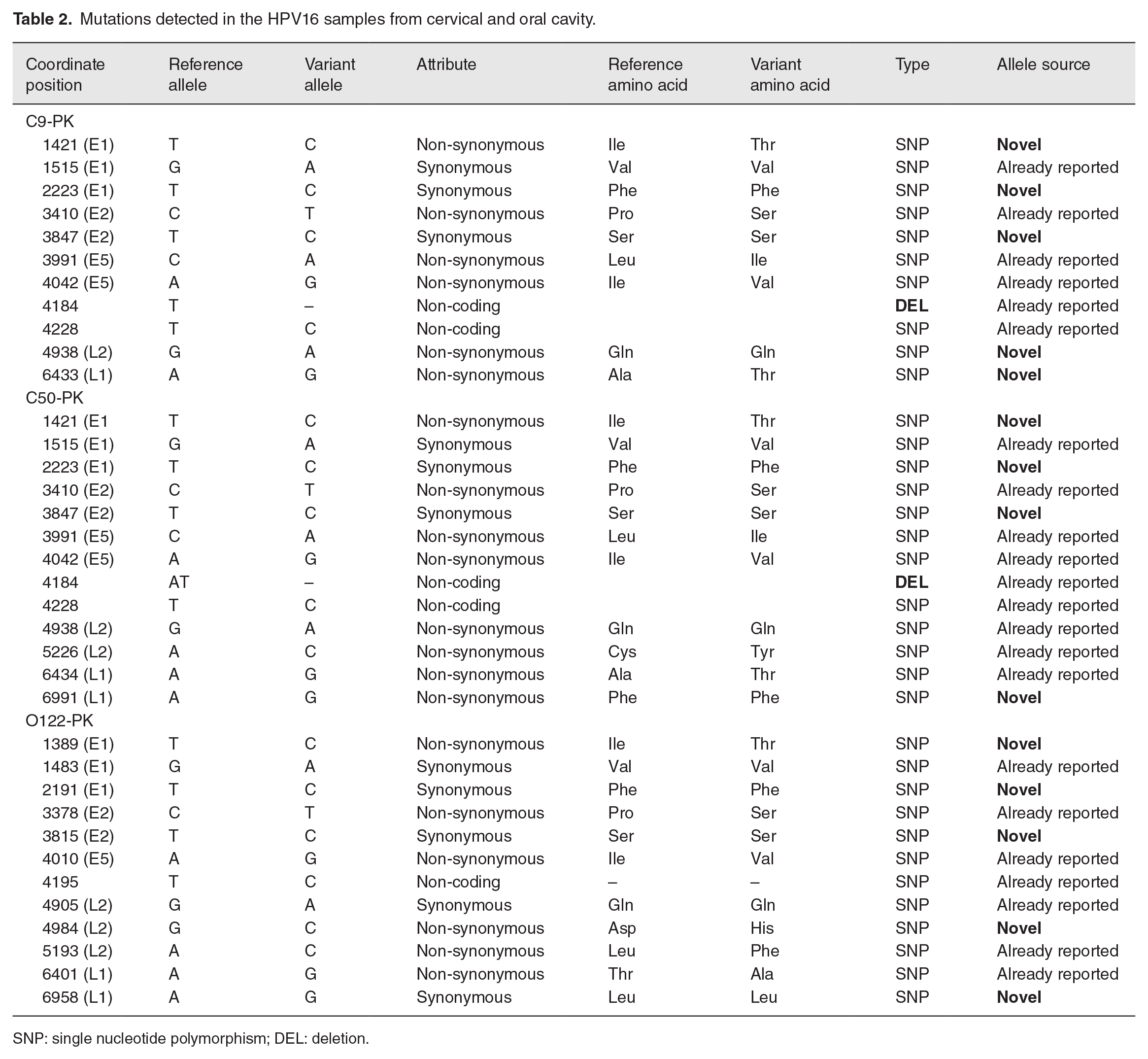
SNP: single nucleotide polymorphism; DEL: deletion.
Variations found in the second cervical sample (C50-PK) contain 13 SNPs in both coding (11 SNPs) and non-coding regions (two SNPs). In the non-coding region, there was only one deletion (AT). The majority of nucleotide variations were detected in the E1 area, followed by the E2, L2, and L1 regions which show that variations are equally distributed throughout the genome. Majority of these mutations were non-synonymous (n = 8) with T > C (4/13; 30.7%) being most abundant followed by A > G (3/13; 23.07%) and G > A (2/13; 15.3%), respectively. Four novel mutations have been reported that have not been described earlier in any database (Table 2).
Table 2 shows the mutations noticed in a sample from the oral cavity (O122-PK). Except for one heterozygous mutation, all mutations were homozygous. Twelve nucleotide variations were observed in the sample, seven of which were already reported SNPs, whereas five novel mutations (SNPs) were observed in the coding region. The majority of SNPs were noticed in the coding region. The most frequent variant regions in the oral sample were each E1 and L2 (25%) genes, followed by E2 and L1 (16.6%) genes, respectively. In the assessed O-122 sample, most of the mutations found were non-synonymous lying mostly in the L2 region as compared to synonymous mutations which were seen to occur mainly in the E1 region. Among the novel mutations, the majority were observed to be synonymous and were observed in the E1 region. Among the variations, T- to -C (4/12; 33.3%) were more prevalent followed by A- to -G (3/12; 25%) and G- to -A (2/12; 16.6%), respectively.
The phylogenetic analysis was carried out according to the maximum likelihood method of HPV16 whole genomes including three unique HPV16 genomes recognized in this study and 30 already reported representatives of HPV16 WGS (NCBI accession numbers Table 3). Study sequences were labeled as C9-PK, C50-PK, and O122-PK.
Accession numbers retrieved from NCBI (gene bank) for the phylogenetic analysis of HPV16.

The phylogenetic tree showed that the HPV16 whole genome from this study clustered with other WGS of HPV16 from Thailand, the United States, China, and Europe with a maximum 99.8% nucleotide identity with the United States (AF125673; AY686580; AY686584), Thailand (FJ610147; FJ610149), China (FJ006723), and Germany (NC001526) in the same cluster. The tree showed two distinct clusters of the genomes (Figure 4).
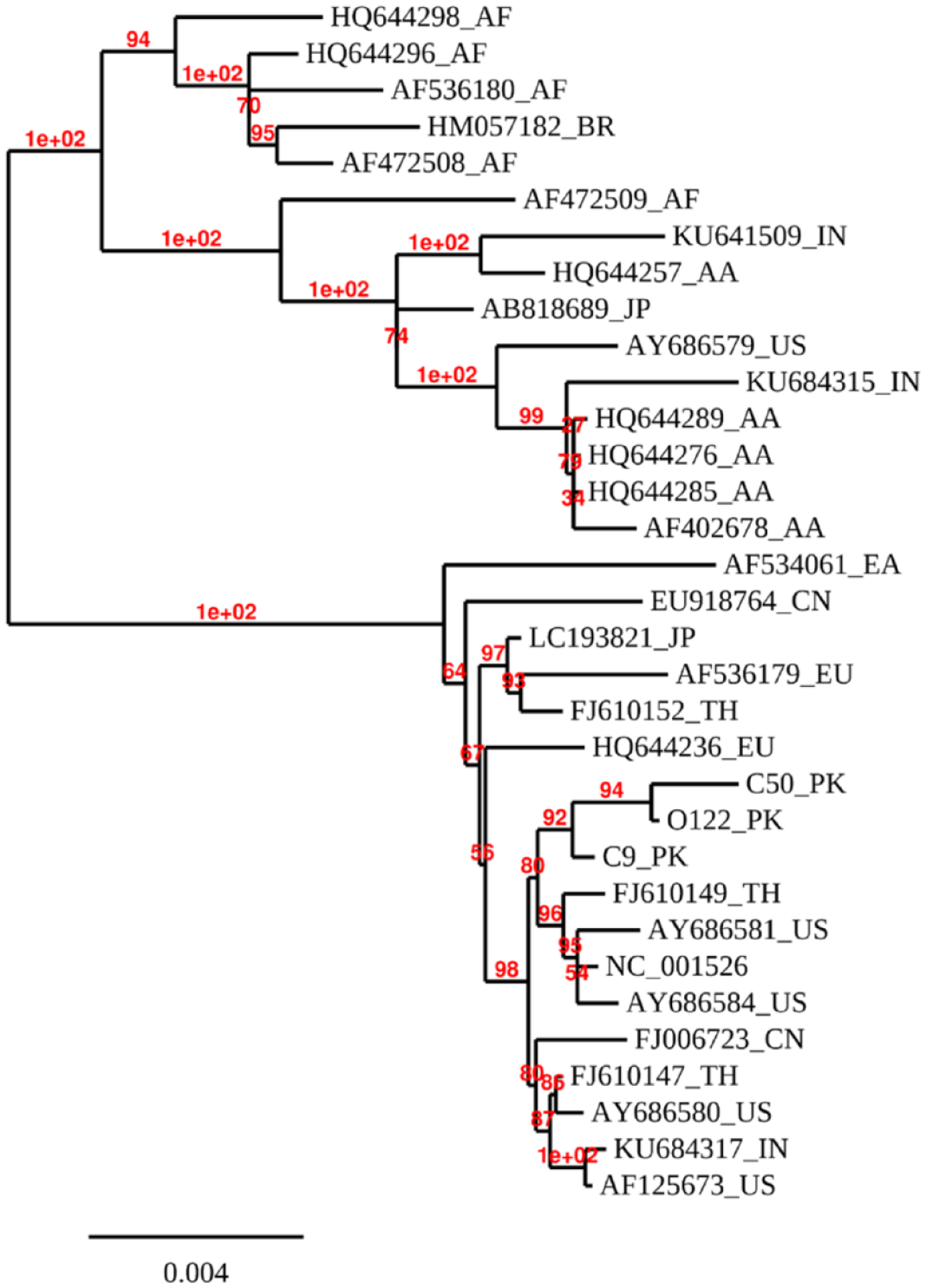
Phylogenetic tree based on WGS analyses of oral and cervical samples positive for HPV16 and sequences from the NCBI gene bank databases.
Discussion
Pakistan is one of the populous countries contributing to the worldwide burden of CC, out of which HPV16 is commonly found in women with cervical and oral cancer worldwide in numerous studies and few HPV16 variants are highly oncogenic in contrast to others.25–27 This is the first national study that used WGS of HPV16, the most frequent HR-HPV type recognized in community-based studies carried out in Pakistan from 2016 to 2021.3,5,18 The study presented here was not designed to evaluate persistence but rather aims to reveal HPV16 genetic polymorphism and some unique HPV16 variants circulating in Pakistani women.
We were able to characterize the genomic variability and evolutionary phylogeny of HPV16 due to our WGS data, which was not possible with limited region sequencing. This enables us to uncover additional SNPs and define HPV lineages and sublineages across the genome using this data set. The distribution of main HPV16 lineages has previously been reported to be demographically conserved,28–32 with historical co-evolution of HPV with humans, as well as more recent migration patterns (particularly from Europe and Africa to the Americas)33,34 thought to be driving factors. In addition, this is the first study that reports the distribution of HPV16 variant lineage/sub-lineages in Pakistan based on the HPV16 complete genome sequences obtained from three Pakistani females without any cervical and oral lesions. The study thus reports HPV16 whole genome from females with gynecological problems rather than already suffering from cervical and oral cancer.
The initial data obtained from our study revealed that samples C9-PK and C50-PK have some common mutations probably due to the reason that both are from the cervical sample while mutations in O122-PK are different because the sample is from a different source, hence showing us that the HPV virus causing different types of cancer has also different SNPs. This has been reported previously in various studies where variants linked with HPV-16 lineage were consistently associated with a higher risk of infection in cervical cancer development.16,35,36 It has been investigated that the association of non-European variants in HPV16 type were even more persistent with the risk of cervical neoplasia as compared to variants linked to European ethnicity. 35 In this study, several new SNPs were recognized in addition to previously reported mutations. A total of 36 SNPs and two indels (insertion/deletions) across the three samples of the HPV16 genome from cervical and oral samples were identified. All mutations were homozygous in cervical and oral samples except one heterozygous mutation that was observed in the oral sample. Regions with a larger number of SNPs were the trans-acting E1 and E2 core proteins, followed by L2 regions which are the targets of neutralizing antibodies, indicating the development of abnormal clones. Previous studies have shown that mutations induced in E1 or E2 regions can increase the HPV 16 genome to immortalize the primary human keratinocytes. 37 Another study on Asian American has found that the presence of variants in transcriptionally active E2 and LTR regions upregulates the HPV 16 promoter activity in cervical cancers. 38 The activation mechanism for development from advanced preinvasive lesions to invasive cervical cancer has also been suggested due to alteration of the E2 gene during viral DNA incorporation into the cellular genome. 14 Previously, it was suggested that the HPV 16 Asian linked variation E232 K in E2 region enhances dose-dependent inhibition of LCR (Long control region) activity and enhances the virus’s ability to induce cancer. 39 Among SNPs present in coding regions, 21 were non-synonymous and 10 were synonymous mutations in all three samples of HPV16. Prominent non-synonymous mutations include the substitution of the T allele into the C allele at position 1421 of the E1 region resulting in the conversion of Isoleucine to Threonine. Pathogenic variations within the HPV16 genome may cause changes in amino acid sequences which may affect the function of each subunit of the virus e.g. changes of L1 could affect the efficiency of infection or later viral antigenicity. However, this does not conclude any change in the biological function of the HPV mode of action because simultaneous variations in several targets change the overall pathophysiology of the disease. 40 The microbiome’s impact on overall health can be also be significant, and has lately been linked to the development of cancer and how well treatments work. 41 Recently 16 S sequencing identifies the abundance of anaerobic bacteria and Lactobacillus iners species in cervical intraepithelial neoplasia and associated with associated with preinvasive disease, increased disease severity, and disease invasiveness. 42 Similarly, patients with HPV-associated oropharyngeal cancer (OPC) and oral cancer (OC) had high levels of Gemella and Leuconostoc, while Haemophilus was associated with HPV infection. The less diversity in microbiome in OC and OPC patients identified through 16 S sequencing indicates that, in contrast to cervical patients, a few dominant, pathogenic bacteria may be involved in HPV persistence and carcinogenesis in the oral environment. 43 These mutations are much more likely in concordance with other studies with similar geographical distributions. 44 E1 is the only enzymatic protein, which assists in the replication of the viral genome inside host cells. 45 Bovine papillomavirus E1 protein has a high degree of sequence homology with HPV E1 protein.46,47 The E1 is a core protein, which results in the formation of a complex with the E2 trans-activator, which is involved in viral replication-related functions such as origin-specific binding and helicase activities. The positions of these variations differ from patients belonging from European or American areas where the majority of the variations are present on E6 and LCR regions, hence indicating that each variant has a different impact on virus persistence and cervical cancer development. 48 The nucleotide diversity analysis of three samples shows that the E5 gene has higher genetic diversity (0.0066) compared to other HPV genes, followed by E2 (0.0018), E1 (0.0015), L2 (0.0014), and L1 (0.0010) genes, respectively. This shows that all through the HPV 16 genome the differences are not equally spread and particularly target HPV genome sub-regions. The newly described variants in HPV16 Pakistani subjects include E1 (1421 T > C), L2 (4938 G > A), L2 (4984G > C), L1 (6433A > G), L1 (6991A > G) and E1 (1389 T > C) non-synonymous variants and E1 (2223 T > C), E1 (2191 T > C), E2 (3847 T > C), E2 (3815 T > C), L1 (6958 A > G) synonymous variants.
To assess the phylogenetic relationship among the Pakistani HPV16 samples, a WGS phylogenetic tree was constructed from cervical and oral samples (C50, C9, and O122) in which two distinct clusters of the genomes were observed. Our results proved that Asian and North American lineages make the mainstream of HPV16 isolates taken from the Pakistani female populations. HPV16 in this study is closely clustered with Thailand (FJ610149_TH) and US genomes (AY686581_US and NC_001526) which show the most likely role of immigrants in the spread of HPV infection throughout the world. The genome also has a close resemblance with Indian, China, and European genomes. The clustering with the Indian genome is because people from both regions shared a common ancestral history and it is a possibility that a shared ancestral linkage with a similar genetic sequence translates into a similar host response toward HPV infection. Our results have been in concordance with a previous study in the Pakistani population where two subjects from Punjab province were amplified and sequenced, thus confirming with HPV16 genotype, and reported phylogenetic association with the isolates from Costa Rica and Japan. 5 The studies carried out in China stated that HPV16 samples were predominantly clustered with the Asian lineages, which is in line with the existing study where the WGS samples were closely related to the Asian lineages.42,43
Despite the identification of several variants and its phylogenetic linkage analysis, the study has some limitations. As the limited number of sequences and participants resulted in insufficient power to investigate the variants, 41 future studies need to investigate the replication of these variants in a large cohort.
RNA sequencing should also be used as a tool for HPV detection since it not only allows for the detection of HPV but also provides additional information related to oncogene expression. A recent study by Song et al. showed several novel HPV16 E7-regulated candidate genes were significantly involved in tumorigenesis process using digital RNA sequencing. 49 Similarly, RNA sequencing along with RT-qPCR provides evidence that 194 Long non-coding RNAs (lncRNAs) were differentially regulated in high-risk (HR) HPV infection along with cervical lesion progression. 50 Furthermore, functional investigations of HPV polymorphisms across the HPV16 genome of South Asian variations should be carried out to investigate DNA profiling of carcinogenicity. In addition, with the rapidly increasing genome sequence data available, the traditional HPV-driven cervical and OC disease mechanisms have to be re-examined. Therefore, it is pertinent to understand how HPV and its various sub-types promote the development of cervical and oral cancers.
In the oral sample, the non-coding region could not be assembled. The same was the case with an independent study where HPV16 collected from an OC sample retrieved the whole coding part but could not assemble the large non-coding region (Kashif; personal communication). This may be due to either the oral samples having a highly heterogeneous sequence that fails to assemble, or whether they completely lack the large non-coding part could be confirmed by single long-range sequencing. These lncRNAs have been previously reported in HPV-associated premalignant lesions and cancers. It was demonstrated that the HPV16 E6/E7 expression deregulates lncRNA expression in normal human epithelial cells and disrupts the key cellular processes.51–53
With the advent of third-generation single-molecule real-time sequencing platforms which are often considered the gold standard in de novo assembly of microbial genomes, complete contiguous genome assembly can be achieved along with identifying complex genomic elements like disease-causing repeat expansions or structural variants.
Conclusions
This study performed WGS HPV16 sequencing analysis from cervical and oral samples obtained from females with general gynecological issues. The nucleotide variation from the WGS of the HPV16 prototype was observed in this study with about 14 newly described non-synonymous and synonymous SNVs recognized. As a result of the use of lineage-specific identification of SNPs or lineage-specific indels detected in short sequence reads in various designated locations of the genome, it is possible to classify and name isolates using entire genome sequences. Facts about multiple HPV16 variants in the population of distinct areas have importance in revealing the carcinogenic process of HPV16 and in establishing preventive and therapeutic vaccines against HPV infection.
Supplemental Material
sj-pdf-1-ebm-10.1177_15353702231211861 – Supplemental material for Whole-genome analysis and evolutionary characterization of cervical and oral human papillomavirus 16
Supplemental material, sj-pdf-1-ebm-10.1177_15353702231211861 for Whole-genome analysis and evolutionary characterization of cervical and oral human papillomavirus 16 by Sadia Minhas, Muhammad Kashif, Haseeb Nisar, Muhammad Idrees and Farheen Ansari in Experimental Biology and Medicine
Footnotes
Acknowledgements
The authors would like to thank the patients who have participated in the study.
Authors’ contributions
SW designed the experiment, sample collection, wet lab analysis and manuscript preparation; MK helped in sample identification, sequencing analysis and proofreading of the manuscript; HN performed data-analysis and prepared the manuscript; MI supervised the manuscript and the critical analysis of the whole manuscript; FA supervised the manuscript, providing intellectual content to the manuscript and advised important shortcoming to improve the overall study. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Data availability
The WGS reported in this study are accessible in GenBank (Study Number, GenBank Accession Number): MZ447799, MZ447800, and MZ447801.
Ethical approval
The study was ethically approved by the University of Lahore Institutional Review Board and ethical committee (IMBB/REG/17189).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.