
Abstract
Piwi-interacting RNAs (piRNAs) are a distinctive category of single-stranded, noncoding RNAs that are crucial for regulating gene expression. Recent studies have shown that piRNAs have a major role in regulating germ and stem cell development, and their dysregulation is connected to various diseases. Consequently, precise identification of piRNA-disease correlations is crucial for understanding disease prognosis and therapy. Establishing the relationships between diseases and piRNAs through experimental research poses challenges and requires a substantial amount of cost and time. Computational approaches offer a promising alternative to mitigate these limitations. This study presents an ensemble approach piR-LGBM that relies on sparse autoencoder and Light Gradient Boosting Machine (LightGBM) classifier to uncover new associations between piRNAs and diseases. The proposed framework generates feature vectors by utilizing the information from piRNA sequences, disease semantics, and currently available piRNA-disease correlation data. The extracted features are then passed through a sparse autoencoder and subsequently to a LightGBM classifier for predicting novel piRNA-disease associations. piR-LGBM yielded an AUC of 0.9640 on fivefold cross-validation. We analyzed the performance of the model against leading methods and classifiers. The empirical results and case-based insights highlight piR-LGBM’s efficacy in identifying piRNA biomarkers behind diseases.
Introduction
Piwi-interacting RNAs (piRNAs) are a unique type of small noncoding RNAs, typically ranging from 23 to 36 nucleotides in length, and are predominantly found in germline cells. They perform biological functions through the interaction with Piwi proteins that belong to the Argonaute protein family (Lin, 2007; Ozata et al., 2019). PiRNAs have a major role in preserving genome integrity by suppressing transposable elements, maintaining gene stability, and contributing to the progression of various diseases (Seto et al., 2007). Although piRNAs were initially identified in germ cells, they have been detected in various somatic tissues as well (Ali et al., 2022; Han and Zamore, 2014). Growing evidence suggests that piRNAs play notable roles in the development of several diseases, such as Alzheimer’s disease (Jain et al., 2019), cancer (Li et al., 2025; Zhao et al., 2025), cardiovascular disorders (Shivam et al., 2025), and immune disorders (Jiang et al., 2024). With the rapid advancements in biotechnology and computational methods (Roy et al., 2017; Zeng et al., 2022), an increasing number of piRNAs have been identified, along with their associated functions. However, the exploration of piRNAs and their roles in human diseases is still in the nascent phase.
Recent advancements in databases such as piRDisease v1.0 (Muhammad et al., 2019), MNDR 4.0 (Chen et al., 2023), and piRTarBase (Wu et al., 2019) that describe the piRNA-disease relationships facilitated research pertaining to piRNAs and diseases. The studies on piRNA-disease association predictions based on computational approaches can be classified primarily into two main types: network-oriented methods and machine learning-oriented methods. In network-oriented approach, piRNAs and diseases are represented as nodes in the heterogeneous network; the intra-edges among piRNAs and diseases represent their respective similarities, and the inter-edges represent validated associations between them (Shi et al., 2024; Zheng et al., 2023). SPRDA (Zheng et al., 2023) constructs a heterogeneous network using similarity information obtained from piRNA sequence data and disease-related functional information and applies a structural perturbation strategy to predict potential piRNA-disease relationships. LKLPDA (Shi et al., 2024) utilizes the LRFKL method, which combines low-rank representation and fast kernel to fuse piRNA and disease similarity matrices and is then given to an open-source machine learning framework for association prediction.
In machine learning approaches, features are constructed using piRNA sequence-level data, disease-related functional data, and empirically proven piRNA-disease associations. The developed features are utilized to train relevant data-driven machine learning models to identify piRNAs behind diseases. iPiDi-PUL (Wei et al., 2021) applies Principal Component Analysis for feature extraction and trains multiple Random Forest models with identical positive and varying negative samples, thus identifying novel associations. iPiDA-sHN (Wei et al., 2020) uses CNN to capture complex patterns from piRNA-disease feature vectors, followed by an Support Vector Machine (SVM) classifier to identify potential associations. MSRDA (Zheng et al., 2022) uses a stacked autoencoder to integrate multiple data sources and identify potential piRNA-disease associations, followed by a Random Forest classifier for prediction. iPiDA-GCN (Hou et al., 2022) constructed graphs, and by using graph convolutional network (GCN), they extracted features and predicted new associations. iPiDA-SWGCN (Hou et al., 2023) extends iPiDA-GCN by addressing sparsity using a weighted association matrix instead of a Boolean one and employs GCN for the final prediction. MambaCAttnGCN+ (Yao et al., 2025) is a computational framework that uses MambaTextCNN to extract piRNA features and a heterogeneous graph convolution method is applied to identify potential associations. PUTransGCN (Chen et al., 2024) uses a heterogeneous GCN with attention to capture key biological features and applies positive-unlabeled learning to handle data imbalance. PPDAMEGCN (Peng et al., 2025) uses a multi-edge-type GCN to integrate similarity information and learn feature representations, followed by a multilayer perceptron for association prediction. iPiDA_CL (Hu et al., 2025) proposes a contrastive learning-based framework for piRNA-disease prediction that eliminates the need for negative samples and improves accuracy and robustness. iPiDA-LGE (Wei et al., 2025) employs a dual GCN framework that integrates local and global piRNA-disease graphs to capture both specific and general features, thereby improving prediction performance.
In this work, we present an ensemble model comprising a sparse autoencoder and a LightGBM classifier to infer novel relationships between piRNAs and diseases. To generate feature vectors, the approach combines sequence-based similarities among piRNAs, semantic similarities between diseases, Gaussian interaction profile (GIP) kernel similarities for both piRNAs and diseases, and known piRNA-disease associations. These feature vectors are then input into a sparse autoencoder, which reduces their dimensionality and extracts the most informative features. The refined features are subsequently used to train a LightGBM classifier, thus predicting the potential piRNA-disease associations along with their corresponding correlation scores. The proposed model achieved an AUC score of 0.9640 in the cross-validation carried out in five iterations using different data splits. We compared the result with previous methods and competing classifiers and conducted case studies on various diseases. The experimental validation and case studies reveal the model’s predictive power in identifying potential piRNA biomarkers.
Materials and Methods
Dataset
This section explains the data utilized in the proposed study.
PiRNA-disease associations
We obtained experimentally validated piRNA-disease associations from the piRDisease v1.0 database (http://www.piwirna2disease.org/index.php), which is used as the benchmark dataset in our study. This database contains 7939 validated interactions between 4796 piRNAs and 28 distinct diseases. To eliminate redundancy, we removed redundant values and associations that are not validated. Finally, we obtained a dataset that contains 5002 experimentally curated piRNA-disease associations, including 4350 piRNAs and 21 diseases. The dataset is represented as follows:

Disease-disease similarity
To calculate semantic similarity between several diseases, this model utilized disease ontology (DO), represented in the form of a Directed Acyclic Graph (DAG) (Nilsson et al., 2021). In this DAG, nodes are used to represent diseases, while edges capture the relationships among them. In addition, the Medical Subject Headings database provides DAG-based hierarchical information for the diseases (https://www.nlm.nih.gov). Using the hierarchical structure of diseases from the DO, the DAG-based algorithm computes interdisease similarity based on semantic relationships, with a higher number of common ancestor nodes indicating greater similarity. The correlation between two diseases
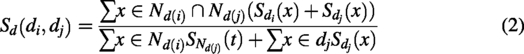
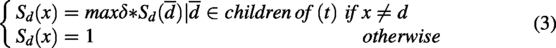
δ indicates the semantic contribution coefficient, which is fixed at 0.5 according to previous studies.
PiRNA–piRNA similarity
The functional similarity among piRNAs
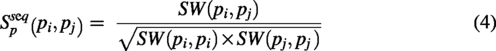
GIP kernel similarity for piRNA and diseases
According to the underlying assumption that functionally similar piRNAs are likely to be linked to similar diseases, and vice versa, GIP kernel similarities were computed for both piRNAs and diseases (Van Laarhoven et al., 2011; Yan et al., 2019). The GIP similarities between two piRNAs,
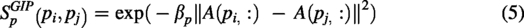
Here,

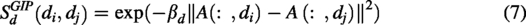
Here,
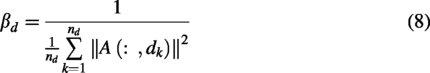
Integrated similarities
The integrated similarities of piRNAs
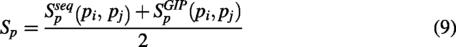
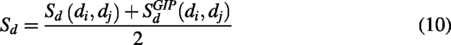
Method
The proposed study piR-LGBM presents an ensemble model that identifies potential piRNA-disease associations. The method comprises two essential parts: Sparse Autoencoder and LightGBM classifier. piR-LGBM utilized pair-wise similarities of piRNAs and diseases and verified piRNA-disease associations for feature construction. For every piRNA-disease pair
Sparse autoencoder-based feature extraction
The autoencoder is an unsupervised neural network architecture capable of capturing high-level features from the input data. A sparse autoencoder is a variant of the standard autoencoder that incorporates a sparsity constraint in the hidden layer, allowing only a small subset of neurons to be active at a time. This constraint is typically enforced by adding a penalty term to the loss function, enabling the model to learn compact and meaningful representations of the input. As a result, sparse autoencoders are well suited for sparse and high-dimensional data and have shown effectiveness in identifying hidden structures in complex biological datasets (Jiang et al., 2020; Liu et al., 2018; Ng, 2011; Sun et al., 2024).
The architecture of a sparse autoencoder includes two core components: the encoder and the decoder. The encoder compresses the input into a reduced-dimensional format, and the decoder works to rebuild the original input from this compact representation. During training, the network updates its weights to reduce the error between the input and its reconstructed version. The basic architecture of a sparse autoencoder is illustrated in Figure 1.

Diagram illustrating the structure of sparse autoencoder.
Typical autoencoders are trained to reconstruct the input data at the output, according to the following equations:.



Here,
In this research, we employed a sparse autoencoder to generate the latent representation of the input vectors. Sparse autoencoders introduce a sparsity penalty that activates only a subset of hidden layer neurons at a time. This penalty term promotes the extraction of highly relevant features from the input data. The objective function used to train the sparse autoencoder is represented as follows:

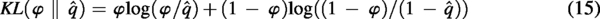
In piR-LGBM, we concatenated the feature vectors of piRNA
LightGBM-based association prediction
LightGBM is a high-performance gradient boosting framework that employs decision tree algorithms to enhance both speed and accuracy, making it a popular choice for classification tasks. It builds a strong predictive model by adding weak learners step by step using gradient boosting. Unlike traditional gradient boosting methods, LightGBM adopts a histogram-based technique and constructs trees by growing leaves leaf-wise instead of expanding them level-by-level, which results in better accuracy and reduced memory usage (Wax and Ziv, 1977). LightGBM has been effectively applied in various biomedical predictive modeling tasks, particularly in identifying associations between diseases and biological entities such as lncRNAs, miRNAs, circRNAs, and microbes, as well as in disease classification and prediction (Ke et al., 2017; Wang et al., 2023; Yang et al., 2024; Zheng et al., 2019; Zhou et al., 2024). In the proposed model, the 128-dimensional samples generated by the sparse autoencoder were used to train the LightGBM classifier to infer the associated interaction score for each piRNA-disease pair in the dataset. The overall concept of piR-LGBM is explained in Figure 2.
The framework of the developed model for the prediction of piRNA-disease associations. The concatenated feature-based representation of piRNAs and diseases is given to the sparse autoencoder, and the resultant condensed features are given to lightGBM classifier for correlation prediction.
Results
Performance evaluation
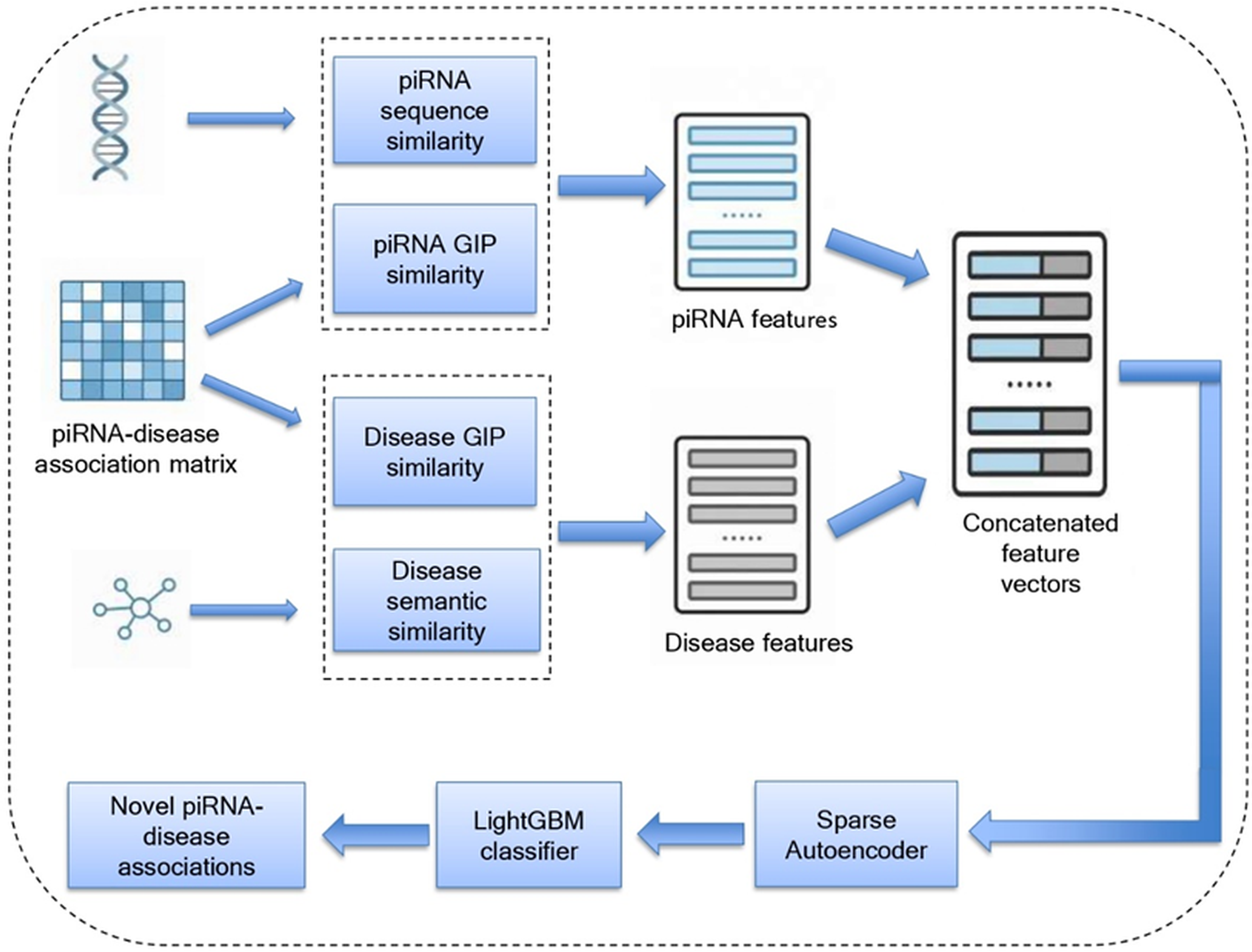
To evaluate the predictive performance of the proposed model, we employed fivefold cross-validation. The dataset was partitioned into five equal subsets, where each subset served as the test set in one iteration, while the other four subsets were used for training. The final result was then obtained by averaging the results across all iterations. The known piRNA-disease associations with prediction scores exceeding the threshold value were classified as true positives (TP), while those falling below the threshold value were identified as false negatives (FN). Likewise, unknown piRNA-disease associations with prediction scores less than the defined cut-off value were grouped as true negatives (TN), whereas those exceeding the threshold value were grouped as false positives (FP). The Receiver Operating Characteristic (ROC) curve was generated by calculating the true positive rate (sensitivity) and false positive rate (1-specificity) across varying thresholds (Figure 3). Here, sensitivity indicates the proportion of TP cases correctly identified, while specificity denotes the proportion of TN cases accurately predicted. The predictive capability of the proposed model was measured by computing the area under the receiver operating characteristic curve (AUC) (Roumeliotis et al., 2024). A higher AUC defines the model’s predictive power in identifying disease-related piRNAs. piR-LGBM gained an AUC score of 0.9640 under fivefold cross-validation. In addition to AUC, we assessed the model’s predictive performance using other evaluation metrics, including accuracy, precision, recall, Area Under the Precision-Recall Curve (AUPR), F1-score, and specificity (Table 1).
ROC curve generated by piR-LGBM during the fivefold cross-validation experiment.
Evaluation Results of piR-LGBM Using Fivefold Cross-Validation

AUC, area under the receiver operating characteristic curve.
Comparative analysis with existing approaches
To assess the predictive capability of piR-LGBM, we conducted a comparative analysis against existing methods predicting piRNA-disease associations. We selected six state-of-the-art models that infer piRNA-disease associations. All these models used the same benchmark dataset, piRDisease v1.0, in their studies. The selected models include MC-GVAE (Sun et al., 2024), PUTransGCN (Chen et al., 2024), piRDA (Ali et al., 2022), iPiDi-PUL (Wei et al., 2021), iPiDA-LTR (Zhang et al., 2022), and PPDAMEGCN (Peng et al., 2025). MC-GVAE used the multichannel graph variational autoencoder to predict the associations. Both PUTransGCN and iPiDi-PUL utilized positive unlabeled learning for the prediction. PUTransGCN combined unlabeled learning with graph convolutional network, while iPiDi-PUL used Principal Component Analysis for feature extraction and Random Forest as the classifier. piRDA used bootstrapping method for the association prediction. iPiDA-LTR used learning to rank technique to predict the piRNAs related to diseases. PPDAMEGCN employed a multi-edge-type graph convolutional network and a multilayer perceptron (MLP) for predicting the piRNA-disease associations. The comparison outcomes are summarized in Supplementary Table S2. The observations highlight the predictive power of piR-LGBM with the state-of-the-art models.
Comparison among different classifiers
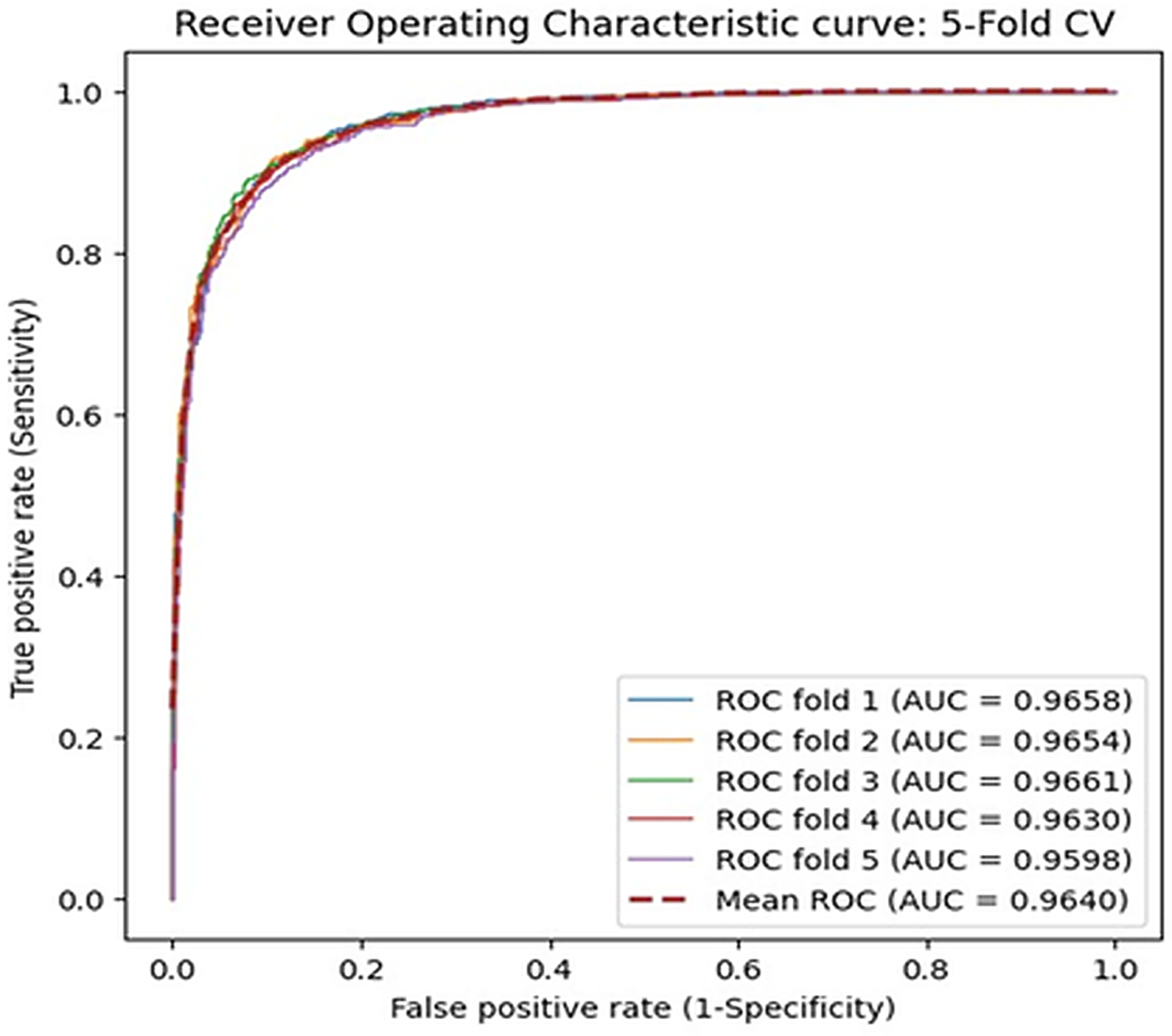
piR-LGBM’s predictive capabilities were further evaluated by comparing it with alternate classification models, including AdaBoost, SVM, and Decision Tree. We applied identical feature construction and feature extraction methods and changed the classifier part only for a fair comparison. We used the same benchmark dataset and assessed the performance through fivefold cross-validations. We obtained AUC scores of 0.9640, 0.9405, 0.9173, and 0.9137 for piR-LGBM, AdaBoost, SVM, and Decision Tree, respectively. The results demonstrated that piR-LGBM outperforms other competing classifiers in predicting piRNA-disease associations. Figure 4 demonstrates the ROC curves obtained by these classifiers.
Comparison with different classifiers: LightGBM, AdaBoost, SVM, and Decision Tree.
Evaluation on an independent dataset
To address the robustness of piR-LGBM on other datasets, we tested it using MNDR v4.0 (Chen et al., 2023), which contains 9616 experimentally validated piRNA-disease associations involving 8205 piRNAs and 15 diseases. The method was assessed using fivefold cross-validation and obtained an AUC of 0.9524, showing reliable performance on a different dataset. We also compared the results with recent methods evaluated on the same dataset. PUTransGCN, PPDAMEGCN, and MambaCAttnGCN+ reported AUC scores of 0.930, 0.931, and 0.940, respectively. Compared with these methods, the proposed model achieves higher AUC on the same dataset. Earlier approaches based on MNDR v2.0 and v3.0 were not included in this comparison due to differences in dataset versions. The ROC curve is given in Figure 5.
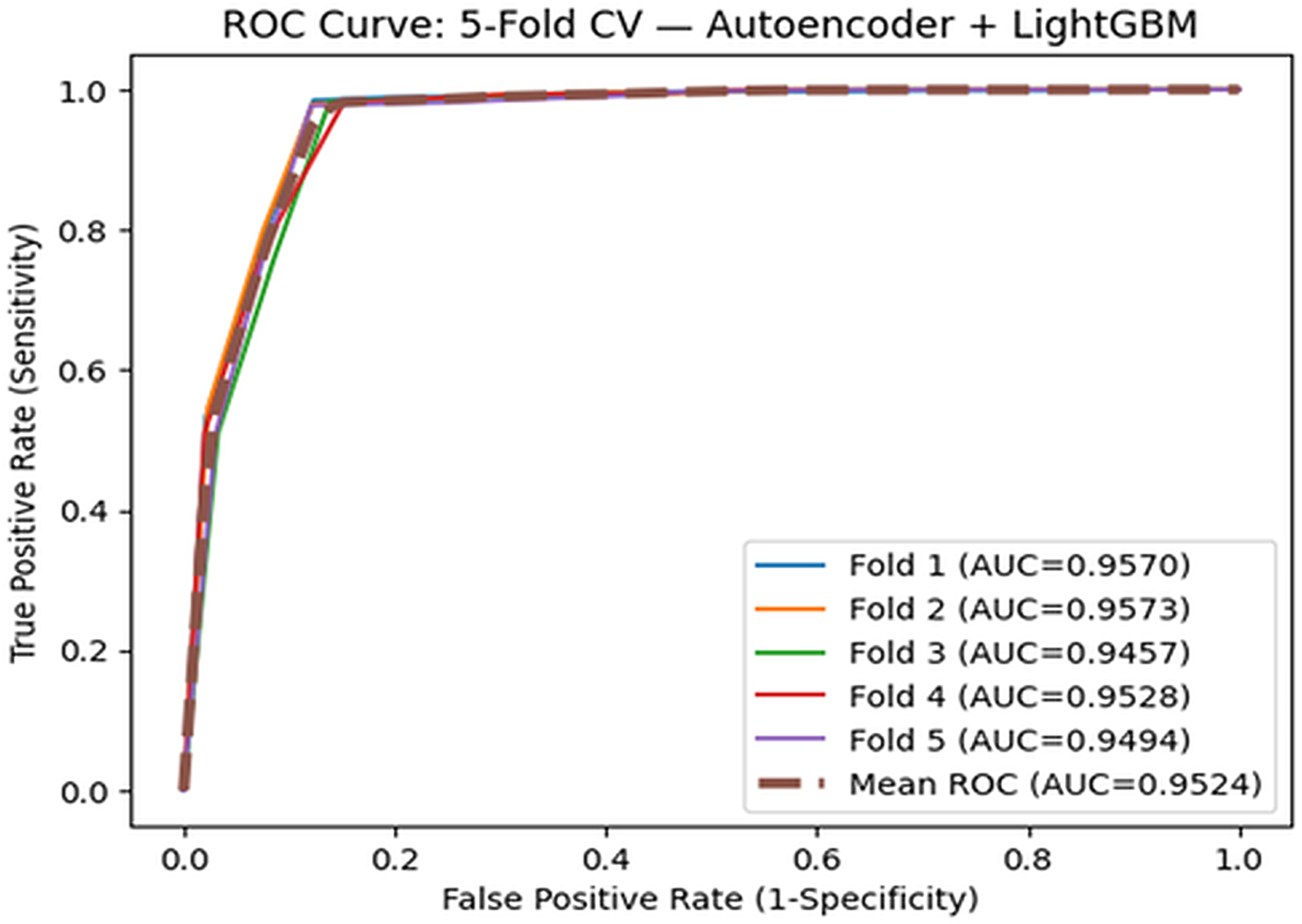
Comparison of the proposed model with MNDR v4.0 dataset, based on fivefold cross-validations.
Ablation experiment
PiR-LGBM includes a sparse autoencoder for feature extraction and a LightGBM classifier for prediction. To assess individual classifier contributions, fivefold cross-validation-based ablation analysis was performed on the benchmark dataset. Initially, the model used LightGBM (AE + LGBM). Subsequently, LightGBM was replaced with AdaBoost (AE + ADB), Support Vector Machine (AE + SVM), and Decision Tree (AE + DT), while retaining autoencoder-based feature extraction. This comparison evaluated each classifier’s impact on performance. Results, including accuracy, sensitivity, specificity, F1-score, AUC, and AUPR, are presented in Figure 6. The findings show that the LightGBM-based model outperformed others, highlighting its effectiveness in handling complex piRNA-disease data.
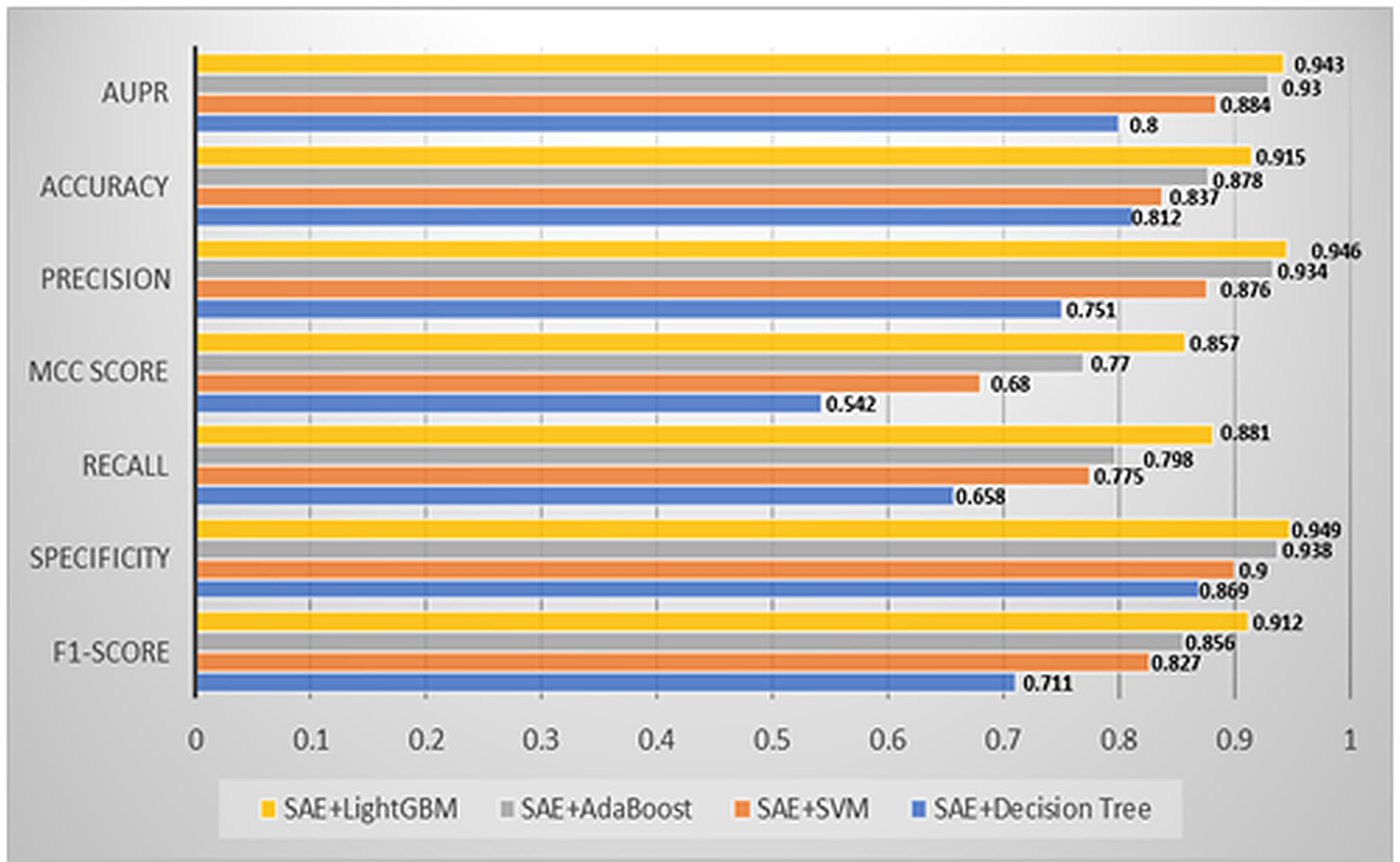
Ablation experiment analysis of piR-LGBM on piRDisease v1.0 dataset.
Case study
Case studies on head and neck cancer, breast cancer, Alzheimer’s disease, and gastric cancer were conducted to validate the model. The model, trained on known associations, generated scores for all unconfirmed piRNA-disease pairs, ranked them in a descending order, and identified the top five piRNAs for each disease (Supplementary Table S3). Of 20 top-predicted piRNAs, most of the associations have been experimentally verified by the latest literature. In head and neck cancer, the upregulation of four piRNAs such as piR-hsa-28190, piR-hsa-28187, piR-hsa-28395, and piR-hsa-28394 and the downregulation of piR-hsa-23655 were observed (Krishnan et al., 2017; Zou et al., 2016). In breast cancer, piR-hsa-1282 and piR-hsa-27493 were found to be upregulated, while piR-hsa-30293 and piR-hsa-26527 were downregulated (Koduru et al., 2017). For Alzheimer’s disease, piR-hsa-1849 was significantly downregulated, whereas piR-hsa-23210, piR-hsa-15023, and piR-hsa-1191 were observed to have increased expression levels. In gastric cancer, piR-hsa-1077 was upregulated in both tissues and cell lines. In addition, piR-hsa-23210 and piR-hsa-26441 were identified as strongly related to gastric cancer. The case study demonstrates the model’s capability to identify piRNA biomarkers behind specific diseases.
Discussion
The framework includes three components: multisource feature construction, sparse autoencoder-based feature extraction, and LightGBM classification. As experimental validation of piRNA-disease associations is costly and time consuming, this approach offers an efficient computational alternative for identifying novel associations. There are several factors that contribute to the efficiency of the model. Ensemble methods tend to outperform individual classifiers by integrating the outcomes of multiple learning algorithms. However, noise in input data poses a significant challenge for accurate classification. Sparse autoencoders effectively address this issue by learning compact representations, denoising the input space, and extracting the most effective features from traditional input feature vectors; this is achieved through a sparsity constraint that enables the model to extract only the most essential features. Training with the reduced dimensional features improved model performance while lowering computational costs.
Similarity computation has a vital role in enhancing the performance of association prediction framework. PiRNA sequence similarities and disease semantic similarities can provide a deeper representation of piRNAs and diseases. The use of GIP kernel similarities helps assign a similarity score to each piRNA-disease pair in the dataset, thereby enhancing the prediction power of proposed model. The major limitation of network-based approaches is that they need to be rebuilt from scratch when new piRNAs or diseases are discovered. Network-based methods fail to predict associations for piRNAs without known disease associations. In contrast, piR-LGBM can handle piRNAs and diseases with no association data. Like other machine learning models, the proposed method is highly adaptable and can easily incorporate new piRNAs, diseases, and piRNA-disease associations into the dataset after similarity computation.
However, the proposed approach has some limitations. It requires both positive and negative samples for training, but obtaining TN samples is challenging or even impossible. To address this issue, negative samples were generated by randomly selecting unconfirmed piRNA-disease associations from the dataset. Although randomly selecting unlabeled piRNA-disease pairs as negative samples is a standard way, some of these pairs may correspond to undiscovered true associations. Future studies could address this limitation by adopting positive-unlabeled learning strategies. Although the proposed model demonstrates strong predictive performance, the dataset used in this study is relatively limited in size and lacks sufficient disease diversity, which may affect its generalizability. Future work can focus on incorporating larger and more diverse datasets to further enhance the robustness and applicability of the model. Furthermore, the current study is limited to human piRNA-disease associations and has not been evaluated on other species such as mice due to the lack of suitable datasets. Future work can be extended to cross-species prediction, particularly in mouse models where abundant cancer-related datasets are available to further assess its robustness and generalizability. In addition, employing more advanced similarity calculation techniques and incorporating diverse biological information, such as piRNA functional and expression profile similarities, into the feature set may further enhance the model’s predictive capabilities.
Conclusion
Understanding disease mechanisms at the molecular level is vital for diagnosis and therapy. Increasing research indicates that piRNAs are strongly linked to human diseases. Identifying disease-associated piRNAs provides insights into molecular mechanisms. This study develops a predictive framework integrating a sparse autoencoder and LightGBM to identify novel piRNA-disease associations. The approach uses piRNA sequence similarity, disease semantic similarity, and known associations to construct features. A sparse autoencoder extracts key representations, which is used by LightGBM to predict novel associations. The framework shows strong potential in identifying disease-related piRNAs. Case studies on head and neck cancer, breast cancer, Alzheimer’s disease, and gastric cancer support predicted associations. Overall, results demonstrate the framework’s effectiveness as a computational tool for uncovering novel relationships and potential biomarkers. With growing experimental datasets, predictive accuracy is expected to improve, and the framework can be extended to other biomolecule-disease associations.
Conflict of Interest
The authors declare that they have no conflict of interest.
Informed Consent
Informed consent has been derived from all the participants.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.