
Abstract
Cancer stem cells (CSCs) are a small population of cells within a tumor which perform stem cell like functions including self-renewal, differentiation and initiation. This narrative review aims to provide an overview of the role of CSCs in resistance to standard treatment and metastasis in breast cancer, potential treatment strategies including latest clinical trials, and the clinical implications for CSCs in the management of breast cancer. The most established breast CSC markers (CD44+/CD24- and ALDH1) are most prevalent in basal-like cancers, the most aggressive subtype of breast cancer. The most implicated CSC pathways include Hedgehog, Notch, CXCR1, and Wnt. Previous phase I/II trials targeting these pathways associated with breast CSCs have shown mixed results; no CSC-specific drug has reached phase III trial in breast cancer. To maximize the potential benefit of CSC-targeted therapy, it would be important to select or enrich patients with CSC markers or CSC-associated pathway activation. It would also be critical to develop combination strategies that overcome plasticity and/or immune invasion, and preferentially destroy CSCs with limited impact on healthy stem cells. Overall, breast CSCs may have greater clinical implications but require further research and development to realize their full clinical potential.
Introduction
Approximately 20 million new cases of cancer are diagnosed each year. 1 Despite advances in surgery, systemic treatment and radiotherapy, many cancers metastasize and/or become resistant to treatment. Breast cancer is the most commonly diagnosed cancer in females worldwide and the leading cause of female cancer death in 112 countries. 1 Despite advances in treatment, many patients with breast cancer still develop recurrence. Patient-level meta-analyses by the Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) demonstrate a 10-year absolute risk of breast cancer recurrence rate of 14.7% in estrogen-receptor positive early stage breast cancer with aromatase inhibitor, 2 and 22.9% in HER2-positive breast cancer with trastuzumab. 3 Despite advances in neoadjuvant chemotherapy and immunotherapy in triple negative breast cancer, about 35% of patients do not achieve pathologic complete response, 4 implying treatment resistance. Thus, it is imperative to identify the mechanisms of cancer treatment resistance and metastasis. The objective of this narrative review is to provide an overview of the role of cancer stem cells (CSCs) in resistance to standard treatment and metastasis in breast cancer. Specifically, this review advances the field by focusing on latest clinical trials and the clinical implications for CSCs in the management of breast cancer.
CSC model
CSCs are a small population of cells within a tumor which perform stem cell like functions including self-renewal, differentiation and initiation.5–9 Although the literature tends to interchange CSCs and tumor initiation cells (TICs), there is a clear distinction but some overlap. TICs are defined as cells which have the capability to form a tumor when transplanted into an animal model (e.g., mouse), whereas CSCs are defined as a subpopulation within a tumor that possess stem-like properties. 10 Stemness defines the intrinsic properties of breast CSCs to self-renew (not a cell cycle state), whereas quiescence is a reversible state of cell cycle arrest (G0) used by CSCs to survive cytotoxic therapy. 11 Dormancy is a prolonged, reversible, non-proliferative state enabling late/metastatic relapse. 11
The theory of CSC was first proposed by Julius Cohnheim in 1870 where he theorized that cancer originated from embryonic-like-cells. 12 However, this theory in the modern era has failed to gain traction due to controversies. The first controversy being that current stem cell markers are generally unreliable and vary between papers/studies. The second controversy is the uncertainty over how CSCs are formed. The traditional model describes a hierarchy in which CSCs sit at the top having the unique ability to differentiate into other tumor cells and self-renew. 13 Since 2020, CSC concept has shifted from a hierarchical model to a plasticity model, recognizing that CSCs are induced through epithelial-to-mesenchymal transition (EMT), which can be reversed by a mesenchymal-epithelial transition (MET).14–16 This model change has implications in the clinical management of breast cancer. Under the previous hierarchical model, the rate of growth was considered predictable. The new model, however, recognizes that CSCs are highly dynamic, shaped by cues which even allow non-CSCs to repopulate the CSC pool. This has shifted the focus of therapy from targeting CSCs alone to also blocking state transitions and modifying microenvironments. 17
There are two CSC states: epithelial-like and mesenchymal-like. Epithelial-like breast CSCs are ALDH+, highly proliferative and generally are found in the interior of tumors, driving tumor growth. Mesenchymal-like breast CSCs are CD44+/CD24-, invasive, and aid in tumor migration, dissemination, and therapy resistance. 18 CSCs transition between these two states reversibly. The EMT transition can be induced by cytokines and growth factors (e.g., TGF-β, IL-6, and TNF-α) in the tumor microenvironment,8,19 as well as Notch, Hedgehog and Wnt signaling pathways. 18 The innate acidity of early breast tumors also promote EMT as an adaptive response, enhancing breast CSCs and mesenchymal-like states. 20 Finally, the ER α signaling suppresses EMT, so alteration or loss of signaling can promote EMT. 21 Bone morphogenetic protein (BMP) and HER2 signaling promote MET. 18 Recently, an integrated single-cell RNAseq atlas of >600,000 cells across 138 breast cancer patients identified that different CSC states exist on a continuum, with tumor cells primarily segregated by stemness, epithelial-mesenchymal plasticity (EMP) and basal phenotypes. 22
Despite the controversies, there are potential implications of CSCs in clinical settings. Studies using xenografts models found that CSCs are resistant to conventional treatments such as chemotherapy and radiotherapy and potentially even being enhanced or “activated” by such methods.23,24 Damaged DNA of CSCs activates anti-apoptotic signaling pathways such as the NF-κB signaling pathway that attenuates apoptosis and enhances survival.
23
Other common mechanisms include DNA repair pathways (ATM/ATR, CHK1/2, homologous recombination), activation of Wnt/Hedgehog/Notch signaling pathways, remaining in dormant/quiescent states to avoid targeting, EMT, upregulation of drug efflux pumps (ABCG2 and MDR1), and tumor niche protection (Figure 1).8,25–27 Thus, reappraising current makers and redefining them could lead to greater clinical implications related to cancer initiation, survival and remission. Developing therapies that eradicate CSCs without affecting healthy stem cells could potentially improve patient outcomes. Mechanisms of tumor resistance to chemotherapy and radiotherapy. The diagram summarizes common cancer stem cell-mediated mechanisms including epithelial-mesenchymal transition, enhanced DNA-damage responses, drug efflux, quiescence, protective niche signaling.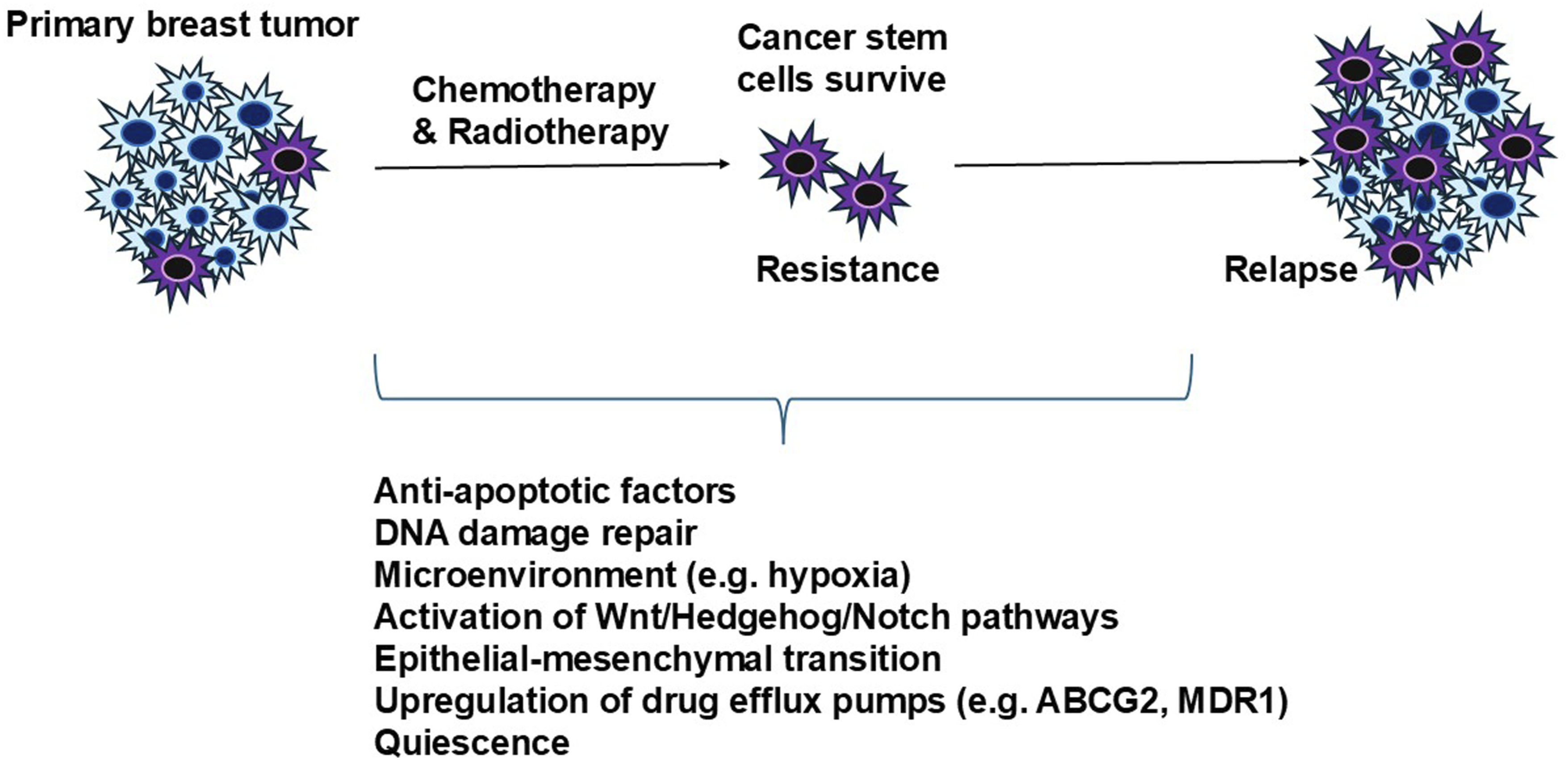
Breast CSC phenotypes and markers
There are two main methods to identify CSCs. The first is to detect these CSCs via their surface markers, for specific molecules such as proteins and carbohydrates. To identify these macromolecules, immunodetection via flow cytometry is often utilized to physically isolate a population of cells with these enrichment markers. 28 Currently, the most established enrichment markers (phenotypes) for breast CSCs are CD44 (+)CD24 (-/low) and ALDH1 (high). 29 Studies have shown that the expression of CSC markers vary across molecular subtypes of breast cancer, and that more aggressive molecular subtypes of breast cancer such as basal-like or TNBC may have a higher percentage of cells with CSC phenotype.29,30 In a study that analyzed 466 invasive breast carcinomas and eight breast cancer cell lines, most of the basal-like tumors (76.5%, 52/68) were of CD44+/CD24- phenotype (defined as ≥ 10% of tumor cells expressing CD44+/CD24-). The CD44+/CD24- phenotype was less frequently observed in luminal A (43%, 127/295), luminal B (41%, 17/41) and HER2+ (27%, 9/33). The ALDH1+ phenotype was also mostly found in basal-like tumors (19%, 13/68), and less abundant in HER2+ (12%, 4/33), luminal B (10%, 4/41) and luminal A (4%, 12/299) breast cancer. 29 Functional markers reflect the activity of CSC, such as ALDH1 enzymatic activity assay, and mammosphere-forming efficiency. Enrichment and functional markers capture overlapping but distinct CSC subpopulations that are dynamic, and shaped by EMT/MET state and surrounding microenvironment of the tumor subtype. 31 Consequently, no universally specific marker exists, and most CSC markers are also expressed by normal tissue stem cells.
Breast CSC signaling pathways
Signaling pathways targeting breast cancer stem cells.
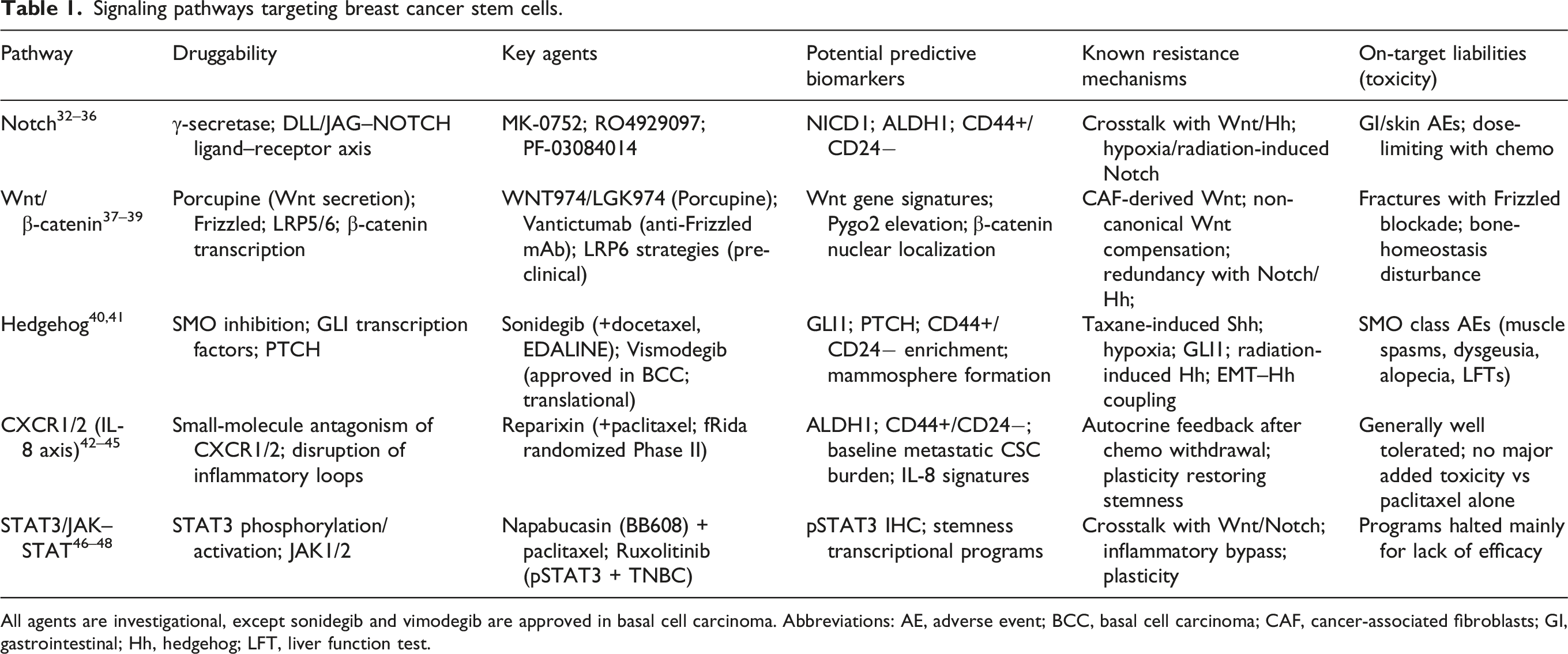
All agents are investigational, except sonidegib and vimodegib are approved in basal cell carcinoma. Abbreviations: AE, adverse event; BCC, basal cell carcinoma; CAF, cancer-associated fibroblasts; GI, gastrointestinal; Hh, hedgehog; LFT, liver function test.
The Hedgehog pathway normally regulates cell proliferation, organ repair, and tissue structure.49–52 Within CSCs, the Hedgehog pathway is essential for the maintenance and proliferation of the CD44+/CD24-(low) population, which have been shown to be resistant to paclitaxel chemotherapy. 53 Hedgehog pathway has also been implicated in providing stem-like features, promoting EMT, angiogenesis, and resisting conventional therapies.54–57 The expression of Patched receptor (PTCH, a key component of the Hedgehog pathway) and the CD44+/CD24- phenotype have both been found to be independent prognostic factors for decreased disease-free survival in 266 patients with breast cancer. 56 Hedgehog was also found to be related to mammosphere development, initiation, and enhancement of markers (phenotypes such as mammospheres, CD44+/CD24-, and ALDH1) (leading to resistance/recurrence). 58 For example, a study found that Sonic Hedgehog (Shh) increased primary mammosphere (when mammary stem cells aggregate) formation by 57% and the average cell number in the mammospheres by 62%. 59 Conversely, cyclopamine, an inhibitor of the Hedgehog pathway decreased mammosphere formation by 45%. 59 Interestingly, Shh is released from breast cancer cell lines following exposure to docetaxel chemotherapy, and activation of the Hedgehog signaling pathway is required for breast CSC survival and expansion after chemotherapy treatment. 54 Following conventional radiotherapy treatment, Hedgehog pathway transcription factor Glioma-Associated Oncogene 1 (GLI1) activates RNA polymerase I in response to radiotherapy, resulting in the emergence of a radioresistant tumor population. 60
Typically, the Notch pathway regulates proliferation and maturity of adult beta cells, differentiation, regeneration, and homeostasis of T cells and stem cells.61–63 In the context of CSCs, the Notch pathway contributes to the maintenance of CD44 (hi)/CD24 (lo) cells (CSC phenotype). 64 Notch1 knockdown reduced the expression of CD44 (hi)/CD24 (lo) phenotype from 90% to 70% of the cells in the population. 64 Notch1 silencing via short hairpin RNA (shRNA) reduced cellular growth and Matrigel invasion by 60%, and cells transfected with shRNA formed fewer metastases in the brain. 64 Notch signaling is also associated with ALDH1 activity.65,66 In addition, the Notch pathway upregulates the expression of Zinc finger E-box binding homeobox 1 (Zeb1), which maintains stemness, tumor aggressiveness, and interactions between CSCs and adjacent endothelial cells. 67 Furthermore, Enhancer of Zeste Homolog 2 (EZH2), which is overexpressed in TNBC, expands breast CSCs through the activation of Notch1 signaling. 66 EZH2 knockdown reduces CSC frequency, tumor growth, and stem-like phenotypes in breast cancer. 66 Out of the 58 TNBCs in the same study, 83% (48 cases) were EZH2high, with 73% (35/48) of those EZH2high tumors being NOTCH1 intracellular domain (NICD1) positive. 66 Radiation treatment actually activates the Notch pathway, causing CSC enrichment and hypoxia promotion, and therefore radioresistance.68–70
Normally, CXCR1 (interleukin 8 [IL8] receptor) is involved in guiding neutrophil chemotaxis which is used as an immunoresponse against infections and inflammation.71–74 CXCR1 in the context of CSCs promotes inflammation, invasiveness/metastasis, therapy resistance, and self-renewal,75–77 being directly related to the ALDH1 phenotype. 78 A study from 33 cell lines showed that ALDH1+ cells had a 7- to 8-fold increase in CXCR1 expression compared to ALDH1- cells. 78 The same study also found that IL-8 increased invasion of ALDH1+ cells, and that ALDH1+ cells have increased metastatic potential to the bone, lung, soft tissue, and muscle. 78 Inhibition of CXCR1 resulted in an 8-fold decrease in tumorsphere formation after 3 days 42 Another study demonstrated the role of CXCR1 in conferring chemoresistance. Breast cancer cells pretreated with paclitaxel formed ∼2–3 folds more mammospheres and also generated larger spheroids than control, via tumor-cell autocrine secretion of inflammatory cytokines after drug withdrawal. 79
The Wnt pathway is important in normal stem cell survival through its self-renewal, initiation and differentiation properties.80–82 In the context of CSCs, Wnt enriches CD44+/CD24- and ALDH1+ populations, and induces metastasis and mammosphere formation.83–86 Wnt signaling is significantly increased in breast CSC-enriched populations compared to normal stem-like cells. 84 In a study that knocked down the Wnt gene, the apoptotic rate of the cell population increased from 1.17% to 14.5%. 86 The ability of CSCs to migrate following Wnt knockdown also decreased, supporting the importance of the Wnt pathway in enabling metastasis and self-renewal. 86 Another study that inhibited Wnt signaling in vivo using LRP6 (a cell surface receptor crucial for activating the Wnt pathway) found decreased capacity of breast cancer cells to self-renew and metastasize. 87 The Wnt pathway is activated in chemoresistant breast cancer cells, and Pygo2 (a co-activator of Wnt pathway), is the most upregulated gene in chemoresistant breast cancer cells. 27 Of the 31 relapsed patients with available tumor samples, 48% (15/31) showed an increase in Pygo2 expression and 71% (22/31) showed an increase in MDR1 expression after chemotherapy. 27 In addition to chemoresistance, the Wnt/β-catenin pathway is also involved in conferring radioresistance. 88 Extracellular vesicles from radioresistant triple negative breast cancer cells transfer β-catenin to confer radioresistance and enhance CSC activity of bystanders cancer cells. 89
Compensatory signaling & signaling crosstalk
Compensatory signaling limits the efficacy of single-target strategies, as it reinforces CSC niches, metabolic plasticity, and drug-states. When a pathway is inhibited, CSC niches provide backup cues through stromal cells, immune cells and ECM mechanics that substitute the inhibited pathway. 8 Extracellular matrix stiffens due to increase collagen, laminin, and fibronectin deposition, which reinforces CSC survival and plasticity. Stromal cells support CSC maintenance by releasing extracellular vesicles containing growth factors, metabolic substrates, and miRNAs. 8 Immune cells modulate critical cellular functions such as self-renewal, plasticity, survival, and immune evasion. 8 Additionally, CSCs’ intrinsic metabolic plasticity allows them to switch between oxidative phosphorylation, glycolysis and fatty acid oxidation, to preserve themselves during pathway disruption. 8 Furthermore, drug tolerance occurs by stress-induced transcription, and cytokine feedback. 8 Finally, Hedgehog, Notch, Wnt, and CXCR1 networks share downstream effectors to cross-activate each other, creating functional redundancy. 8
Crosstalk among developmental signaling pathways adds an additional layer of complexity to tumor biology and treatment. The complex interconnections between the various signaling pathways are beyond the scope of this narrative review and only select crosstalks are covered here. Figure 2 shows a schematic of the crosstalk between Notch-Wnt-Hedgehog in the tumor microenvironment. The Hedgehog pathway can induce Notch activity by upregulating the Jagged-2 (Notch ligand).
90
The Hedgehog pathway also inhibits Wnt signaling via upregulation of sFRP-1, which in turn suppresses β-catenin transcriptional activity.91,92 At the same time, Hedgehog activation via Smoothened (SMO) receptor increases β-catenin transcriptional activity.
93
Wnt signaling upregulates Jagged-1 (a Notch ligand), increasing Notch activation.
93
Schematic of the crosstalk between Notch-Wnt-Hedgehog in the tumor microenvironment. The schematic highlights compensatory signaling routes that can substitute for single-pathway inhibition.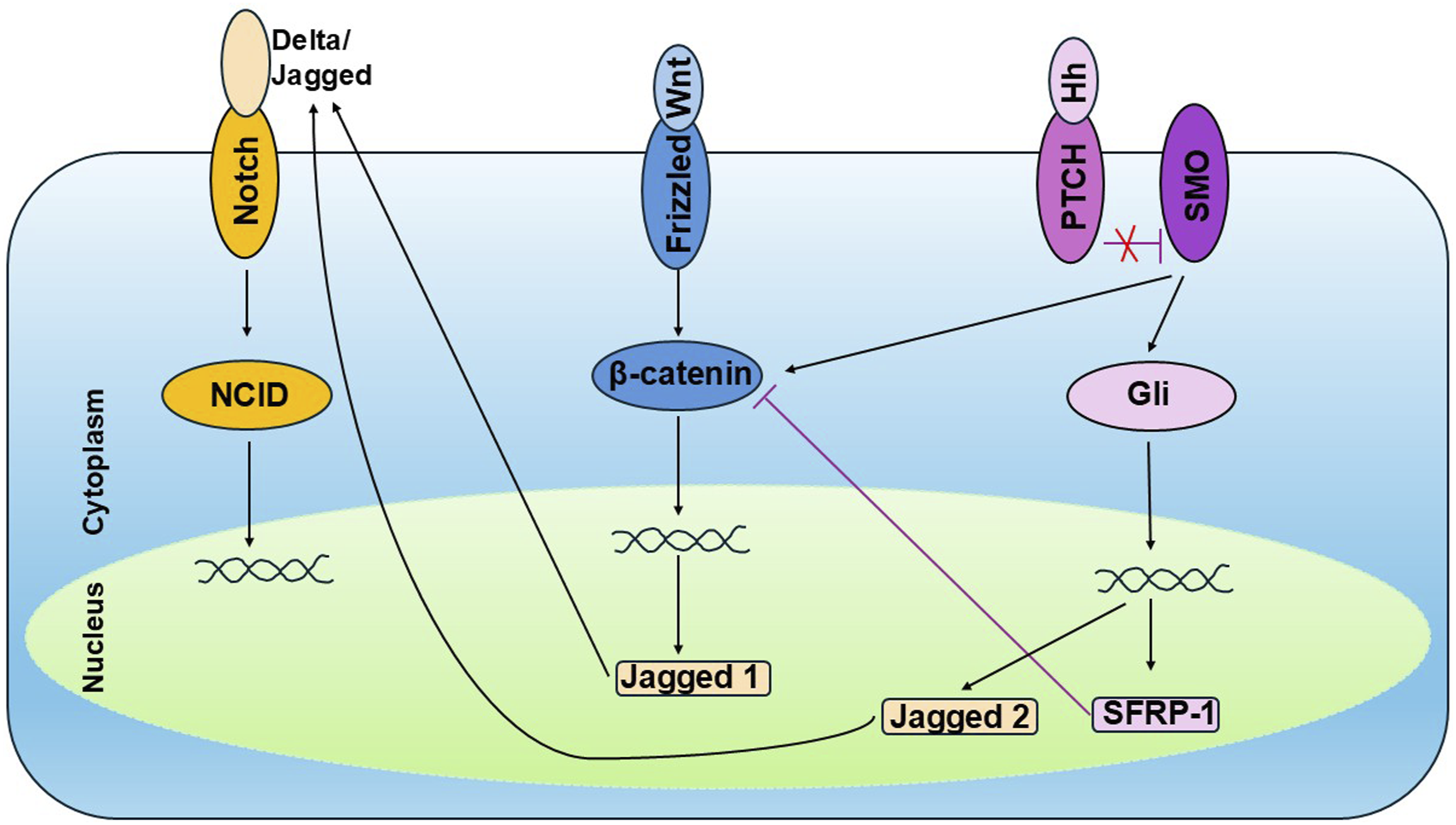
Treatment resistance by molecular subtype
CSCs are also linked to specific resistance patterns in different molecular subtypes of breast cancer. In ER+ breast cancer, CSCs are implicated in resistance to endocrine therapy, due to the enrichment of CD44+/CD24- and ALDH+ populations that survive estrogen deprivation, relying less on ER signaling. 94 Endocrine resistance is driven by JAG1-NOTCH4-dependent CSC activity. 95 In HER2+ breast cancers, expression of CD44+/CD24− (but not ALDH1) is an independent prognostic factor for poor disease-free and overall survival, and a predictive factor of trastuzumab response. 96 Trastuzumab treatment downregulates HER2 expression and leads to an increase in JAG1 expression and subsequent activation of the Notch signaling pathway, which enhances CSC survival. 97 Resistance of TNBC to standard cytotoxic chemotherapy is attributed to inability of chemotherapy to target CSCs, with residual CSCs serving as a reservoir for self-renewal and proliferation. 98 For example, signal transducer and activator of transcription 3 (STAT3) and its downstream pathway, which can convert non-CSCs to CSCs, mediate resistance to doxorubicin in TNBC. 99
Clinical trials targeting breast CSCs
There are currently approved agents specifically for targeting CSCs. However, there are approved agents that target CSC-related pathways. For example, vismodegib and sonidegib are both approved for basal cell carcinoma, and inhibit Smoothened in the Hedgehog signaling pathway. 40 Sonidegib has been shown to downregulate CSC marker expression in TNBC patient-derived xenografts and sensitize breast tumors to docetaxel chemotherapy. 41 In a phase I clinical trial EDALINE of 12 patients with metastatic TNBC treated with sonidegib and docetaxel chemotherapy, one achieved complete response and two had disease stabilization. 41
Reparixin, an inhibitor of CXCR1 (IL-8 receptor), has been shown to selectively deplete CSC population in human breast cancer cell lines and breast cancer xenografts. 42 In a phase IB study, reparixin in combination with weekly paclitaxel chemotherapy appeared to be safe and resulted in a 30% response rate out of 30 patients with HER2-negative metastatic breast cancer. 43 A window-of-opportunity trial in 20 patients with operable HER2-negative breast cancer demonstrated that 21 days of oral reparixin before surgery resulted in a ≥20% decrease in CSC markers on flow cytometry: CD24-/CD44+ and ALDH+ in 9/17 and 4/17 evaluable patients, respectively. 44 Despite these promising results, a randomized phase II trial of paclitaxel ± reparixin in 123 patients with metastatic TNBC showed no difference in progression-free survival (PFS) between the two groups (median 5.5 vs 5.6 months). 45 Possible reasons for the negative results include different dosing regimen in the trial (21 days followed by 7 days off each 28-day cycle vs pre-clinical administration for 28 consecutive days) possibly allowing CSC survival during the break, and uneven distribution of the presence of CSC within the two groups. 45 Based on CSC analysis of metastatic tissue from 54 randomized patients at baseline, more patients in the reparixin arm had CSC, which is associated with worse prognosis (12 vs 4 for ALDH+ and 22 vs 12 for CD24-/CD44+ in reparixin and placebo group, respectively). 45
MK-0752, a γ-secretase inhibitor, was found to reduce breast CSCs by inhibition of the Notch pathway in breast cancer patient-derived xenograft models. 32 It also enhanced the efficacy of docetaxel chemotherapy in vivo. A phase IB trial that included 30 patients with advanced breast cancer and escalating doses of MK-0752 plus docetaxel showed that clinically meaningful and tolerable doses of both drugs were achievable. 32 Of the 24 patients evaluable for response, 11 achieved partial response, nine stable disease and four progressive disease with MK-0752 plus docetaxel. Serial biopsies of patients’ breast tumors showed a decrease in CD44+/CD24−, ALDH+, and mammospheres in serum-free media (MSFE). In a separate phase I study that evaluated MK-0752 and included 24 patients with advanced breast cancer, weekly dosing was generally well tolerated and response was observed in patients with brain tumors (not extracranial solid tumors). 33 Another phase IB study investigated the combination of another γ-secretase inhibitor (RO4929097) with exemestane in 15 patients with estrogen receptor-positive metastatic breast cancer, and found that it was safe and reasonably well tolerated. Of the 14 patients evaluable for response, one achieved partial response, six had stable disease and seven progressive disease. 34 A separate phase I trial studied RO4929097 in combination with neoadjuvant chemotherapy for operable triple negative breast cancer, but did not evaluate anti-CSC effect. 35 A phase I study of another gamma secretase inhibitor, PF-03084014, in combination with docetaxel in patients with advanced TNBC showed limited anti-tumor activity, possibly due to the limited number of patients treated in first-line with the combination at the maximum tolerated dose (MTD). 36 The overall relatively level of anti-tumor activity, which could be related to lack of biomarker selection for Notch amplification/mutation and/or activation of compensatory pathways, and toxicity possibly limiting optimal dosing have halted further development of MK-0752 in breast cancer treatment.
Napabucasin, a first-in-class inhibitor of cancer stemness that targets the STAT3 pathway was administered with weekly paclitaxel in a phase IB/II study in 35 patients with metastatic triple negative breast cancer who progressed on prior systemic therapy. These patients were heavily pretreated, with a median of four prior lines of therapy, including 94% that had progressed on prior taxane-based chemotherapy regimens. Treatment was well tolerated with promising signs of anti-cancer activity: disease control rate 55%, and objective response rate 13%. 46 Unfortunately, the addition of napabucasin to paclitaxel did not improve progression-free nor overall survival in pretreated advanced gastric/gastroesophageal junction adenocarcinoma in a phase III trial. 47 Subsequent efforts on napabucasin were halted. In a phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in 23 patients with pSTAT3-positive metastatic TNBC, no objective responses were observed and the study was closed to further accrual. 48
In a phase I study of bivatuzumab mertasine (which recognizes the variant domain v6 of CD44) in patients with CD44v6-positive metastatic breast cancer, disease stabilization was achieved in 50% of patients. 100 The most common toxicity being mild skin disorders in 75% of patients. As a consequence of one fatal toxic epidermal necrolysis that occurred in an esophageal cancer study running in parallel, the sponsor decided to immediately discontinue clinical trials programme with bivatuzumab mertansine.
Few studies have focused on targeting the Wnt pathway. In a phase 1 study in 94 patients with advanced solid tumors (including 14 with triple negative breast cancer), single-agent treatment with WNT974 (first-in-class Porcupine inhibitor) was generally well tolerated. 37 Porcupine is a membrane-bound O-acyltransferase enzyme that is required for post-translational modification of Wnt ligands in order for them to become functionally active. 38 Analysis of paired tumor samples from the phase I study suggests that WNT974 may promote immune activation in the tumor microenvironment and enhance the activity of checkpoint inhibitors. 37 Another phase IB trial showed the combination of vantictumab (monoclonal antibody that binds Frizzled receptors and inhibits canonical Wnt signaling) plus paclitaxel was generally well tolerated in 48 patients with locally advanced or metastatic HER2-negative breast cancer. 39 The response rate (31%) and clinical benefit rate (69%) were promising. However, six patients experienced bone fractures related to vantictumab day 36–113, likely due to the role of Wnt pathway signaling in bone-homeostasis. The high incidence of bone fracture in this study and other studies 101 limited future development of vantictumab in metastatic breast cancer.
Of the aforementioned pathways and agents, the most promising pathway for therapeutic targeting based on current evidence appears to be the Hedgehog pathway. Hedgehog pathway has been closely linked to chemotherapy and radiation resistance, and FDA-approved hedgehog inhibitor (sonidegib) for basal cell carcinoma also shows activity in breast cancer with manageable toxicity. 41
Ongoing trials targeting breast cancer stem cells.

Clinical considerations and challenges
Despite all of these promising results, why has the theory of breast CSCs not been implemented clinically around the world? This is likely due to the lack of unique and universal CSC markers and pathways, and the difficulty of distinguishing and selectively targeting CSCs. With CSCs being a small part of the tumor population as well as markers varying between patients, it becomes difficult to reliably and routinely detect breast CSCs. Instead of targeting CSC markers, alternative strategies of targeting CSC function include disruption of the CSC niche within the tumor microenvironment, and targeting specific states, metabolic vulnerabilities such as oxidative phosphorylation, or stress-response pathways.
Safety
The development of some inhibitors in breast cancer was discontinued early when studies in other cancers showed excessive toxicity, such as in the case of vantictumab with excessive fragility fractures and bivatuzumab with fatal toxic epidermal necrolysis. The fact that many signaling pathways that regulate CSC self-renewal are also important in normal tissue homeostasis narrows the therapeutic window of CSC-targeted therapies. To limit the impact on normal stem cells, mitigation strategies include local or targeted drug delivery approaches (e.g., intratumoral administration, antibody-drug conjugates) to reduce systemic toxicity, intermittent dosing schedules to allow recovery of normal stem cells, protective co-therapies, selective biomarkers (e.g., stemness gene expression profiles) to identify tumors most dependent on stemness pathways, and FLASH radiotherapy which may protect normal stem cells. 104
Effectiveness
Most of the clinical trials targeting breast CSCs have been conducted in combination with conventional therapies (e.g., chemotherapy), which primarily eradicate rapidly proliferating non-CSC tumor cells but not CSCs. While combination therapy may be more toxic, it still widens the therapeutic window as CSC monotherapy primarily affects self-renewal rather than immediate tumor shrinkage. Although several inhibitors show promising efficacy against CSC in cell lines, animal models and even phase I trials, larger phase II or III trials in humans were negative such as the case with reparixin in combination with chemotherapy. As mentioned above, possible reasons for the negative results include different dosing regimen in the trial than in pre-clinical studies, and uneven distribution of the presence of CSC within the two groups. Many pre-clinical studies use immunodeficient mice with patient-derived tumor xenografts, which may not CSCs also evade the immune surveillance by down-regulation of major histocompatibility complex (MHC)-I and tumor-associated antigens. 105 accurately represent the heterogeneity and tumor microenvironment of human breast cancer. The biologic effects observed in phase I trials (reduced CSC markers and/or response rates) may not necessarily translate into improved survival in phase II/III trials.
Most importantly, CSC plasticity—the ability of non-CSCs to de-differentiate into CSCs under chemotherapy- or radiotherapy-induced stress—explains why trials that target only the baseline CSC fraction frequently fail. This is consistent with the common clinical phenomenon where a tumor initially shrinks (with the sensitive epithelial bulk killed) but later recurs with EMP as a more aggressive, metastatic, and multidrug-resistant mass. At the pathway level, single-node inhibition within the Wnt/Notch/Hedgehog signaling network often results in incomplete suppression because these developmental pathways are redundant and interconnected. Thus, durable clinical responses likely require combination strategies that simultaneously kill rapidly proliferating bulk of tumor cells (with chemotherapy/radiotherapy), 104 and prevent therapy-induced CSC enrichment and repopulation by suppressing stemness/EMT using inhibitors that disrupt interconnected escape mechanisms. However, given that the pathways that sustain CSCs are also involved in normal tissue homeostasis and that combination therapies can cause more toxicity, safety is an important consideration. CSC combination strategies should be implemented rationally via biomarker enrichment (e.g., selecting tumors with evidence of Notch activation for Notch-pathway inhibitors), targeted delivery approaches (e.g., intratumoral delivery), and/or avoiding combination strategies causing additive toxicity. For example, if a CSC inhibitor is known to cause bone fracture (such as in the case of vantictumab), then avoid combining that with high-dose radiation to a weight-bearing bone, which may further increase pathologic fracture risk.
Future directions and conclusion
To maximize the potential benefit of CSC-targeted therapy, it would be important to select or enrich patients with CSC markers or CSC-associated pathway activation. It would also be critical to develop drugs that reach tumor niches where CSCs hide (e.g., hypoxic regions, brain) or drug delivery systems targeting the CSC microenvironment, and preferentially destroy CSCs with limited impact on healthy stem cells. Transitioning from broad inhibitors to targeted approaches will limit the impact on healthy stem cells (Figure 3). Combination strategies to overcome plasticity and/or immune invasion (e.g., checkpoint inhibitors plus stemness inhibitors) may improve treatment response. Finally, personalizing CSC-targeting treatment based on tumor molecular subtyping is key. Overall, breast CSCs may have greater clinical implications but require further research and development to realize their full clinical potential. Transition from broad inhibitors to targeted approaches. The flowchart illustrates therapeutic evolution from broad pathway inhibition to more targeted approaches with emphasis on improving the therapeutic window and minimizing normal stem-cell toxicity.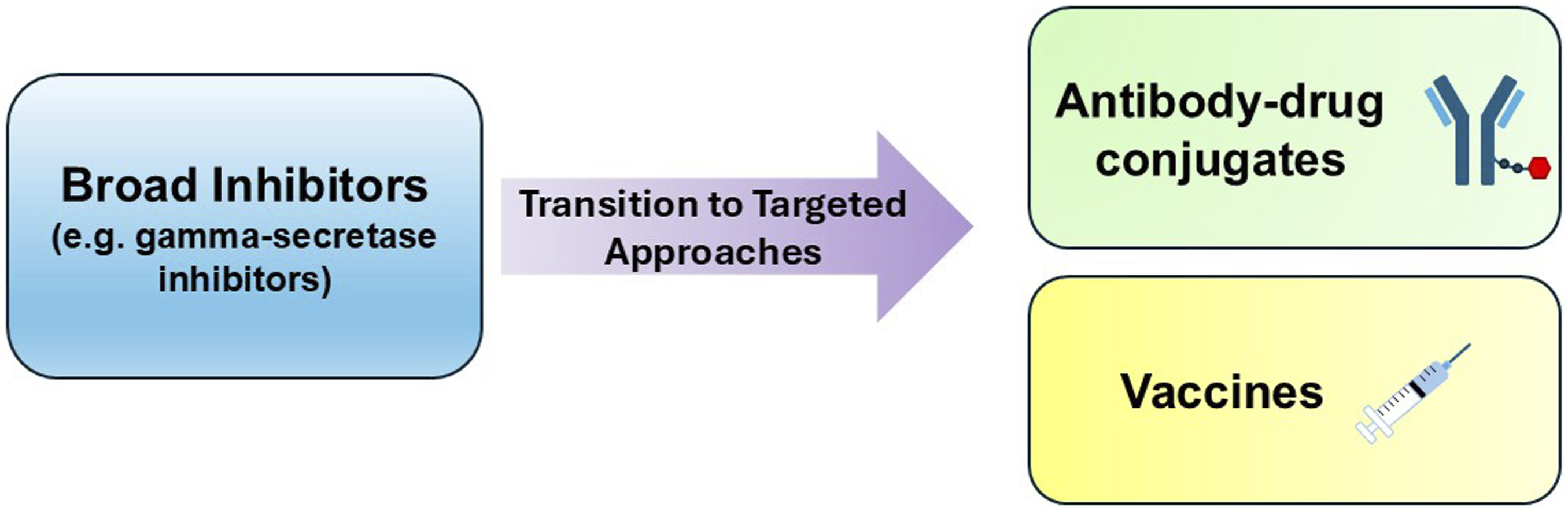
Footnotes
Acknowledgments
The author would like to thank Professor N Landry for her feedback.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.