
Abstract
Background/Objective
Arteriovenous fistulas (AVFs) are the preferred vascular access for hemodialysis of patients with end-stage renal disease. However, there is a high incidence of AVF failures caused by insufficient outward remodeling or venous neo-intimal hyperplasia formation. Abnormal proliferation and migration of vascular smooth muscle cells (VSMCs) play an important role in many cardiovascular diseases. Abnormal VSMC proliferation and migration could be abolished by inhibition of mitochondrial division.
Method
We found that abnormal proliferation and migration of VSMCs and increased mitochondrial fission were associated with AVF stenosis in patients. We also investigated the mechanisms, particularly the role of mitochondrial dynamics, underlying these VSMC behaviors. In vitro, we observed that inhibition of mitochondrial fission and Akt phosphorylation can diminish proliferation and migration of VSMCs induced by platelet-derived growth factor-BB (PDGF-BB). In vivo, daily intraperitoneal injections of mitochondrial division inhibitor 1 (Mdivi-1) decreased VSMC proliferation and reduced AVF wall thickness in a rat AVF model.
Conclusion and Result
Our results suggest that inhibition of mitochondrial fission improves AVF patency by reducing wall thickening through the PI3K/Akt signaling pathway. Therefore, inhibition of mitochondrial fission has the clinical potential to improve AVF patency.
Keywords
Introduction
Arteriovenous fistulas (AVF) are the first choice for patients with end-stage renal disease to create vascular access for hemodialysis. 1 However, only approximately 60% of all AVFs remain patent 1 year after the operation, and the patency rate decreases to 40% after 2 years.2,3 The main cause of AVF failure is venous stenosis of the AVF and ptsubsequent thrombosis resulting from the neo-intimal hyperplastic lesion.4,5 The expansive venous neo-intimal lesion consists of proliferating and migrating cells as well as matrix accumulation, which occurs mainly at juxta-anastomotic sites of the outflow vein proximal to the anastomosis.3,6 However, the mechanisms of venous neo-intimal hyperplasia remain poorly understood.
The thickened intima in the AVF outflow vein is composed primarily of α-smooth muscle actin (α-SMA)-positive cells and scattered inflammatory cells. Neo-intimal hyperplasia can be induced by migration and proliferation of vascular smooth muscle cells (VSMCs) in the intimal layer.7,8 The abnormal proliferation and migration of VSMCs can be stimulated by growth factors and inflammatory cytokines, such as PDGF and TNF-α, after the establishment of an AVF.9,10 Platelet-derived growth factor (PDGF) is a dimeric glycoprotein that can be composed of two A subunits (PDGF-AA), two B subunits (PDGF-BB), or one of each (PDGF-AB). PDGFs are major growth factors that regulate cell growth and division. Among them, PDGF-BB is a crucial mitogen acting on vascular smooth muscle cells. Previous evidence has demonstrated that the levels of PDGF-BB in cardiovascular disease were increased in experimental animal models and patients and that PDGF-BB plays an important role in vascular remodeling.11,12 Therefore, the present study was designed to induce the VSMCs’ proliferation and migration by PDGF-BB.
Mounting evidence indicates that mitochondrial fission accelerates VSMC proliferation after vascular injury.13–15 Thus, mitochondrial fission may contribute to neo-intimal hyperplasia in AVF. Proteins that participate in fission and fusion regulation include dynamin-related protein 1 (Drp1), and mitofusins 1 and 2 (Mfn1 and Mfn2, respectively).16,17 Drp1 is required for mitochondrial fission and acts as a protein marker for mitochondrial dynamics. The contribution of active Drp1 is one of the important factors that affects the function of mitochondria. Strikingly, phosphorylation of Drp1 plays a crucial role in Drp1 activity regulation. 18 Accumulating evidence indicates that Ser-616 phosphorylation promotes the translocation of Drp1 from the cytosol to the mitochondrial outer membrane.19,20 However, little is known about whether inhibition of mitochondrial fission could diminish proliferation and migration of VSMCs induced by PDGF-BB as well as inhibit the formation of stenosis in AVF.
PDGF-BB is among the most potent mitogens that cause the proliferation and migration of VSMCs via activating various intracellular signaling pathways, including the PI3K/Akt, Mtor, and AMPK pathways.21–23
In the present study, we demonstrated that mitochondrial division inhibitor 1 (Mdivi-1), an inhibitor of mitochondrial fission, can attenuate VSMC proliferation and migration, and inhibit neo-intimal formation in vivo and in vitro. We also confirmed that the PI3K/Akt signaling pathway is associated with the progression of neo-intimal thickening.
Materials and methods
Demographic data and comorbidities of patients
Demographic data and comorbidities of patients.
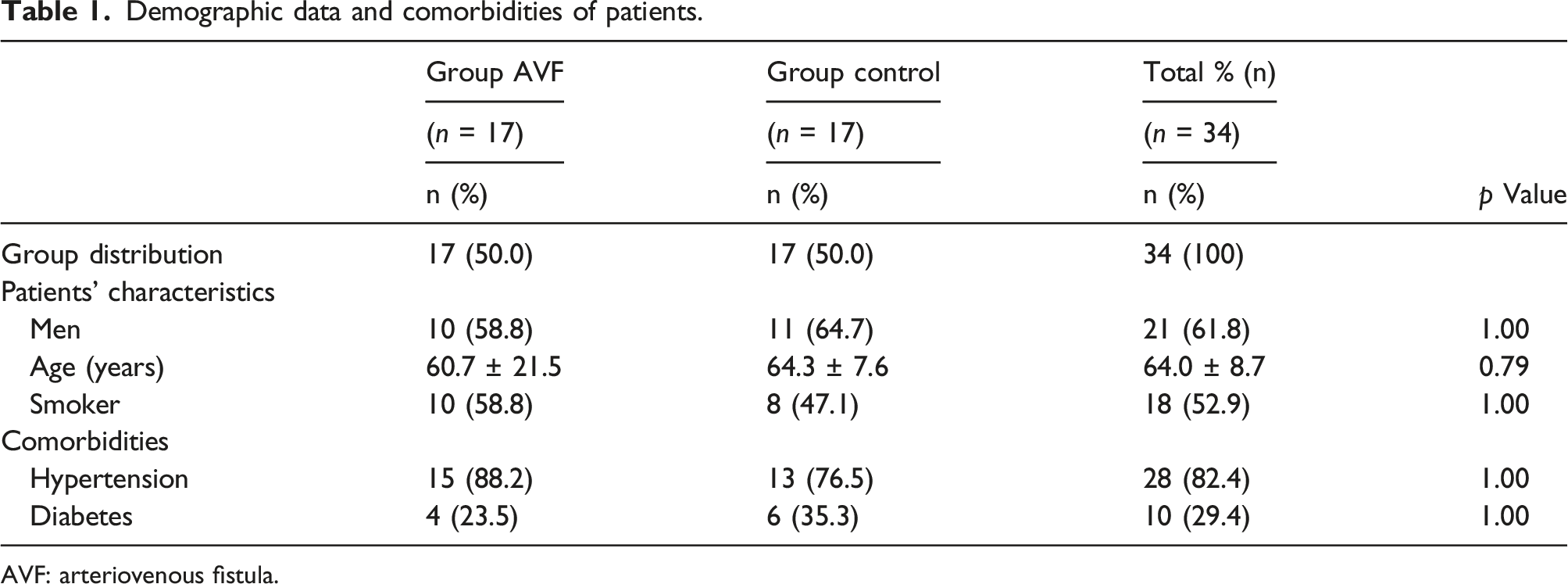
AVF: arteriovenous fistula.
Animal model
Male Sprague Dawley rats weighing 250–300g were purchased from Vital River (Beijing, China). Rats were housed in the Experimental Animal Center of the China-Japan Friendship Hospital (Beijing, China) at a temperature of 25°C, relative humidity of 70%, and a 12/12 h light/dark cycle. All experimental procedures were approved by the Ethics Committee of Animal Research, Peking University Health Science Center, and the investigation conformed to the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). Following 1 week of adapting to their new environment, male rats were randomly divided into two groups: AVF group (n = 10) and sham group (n = 10) to be analyzed on the same schedule. AVF group rats were anesthetized with 75 mg/kg of pentobarbital, and heparin (100 U/kg) was injected via the tail vein to prevent coagulation. After achieving general anesthesia, a 1.5-cm oblique incision was made in the neck slightly off-center, and the right submandibular gland and right sternocleidomastoid muscle were freed and resected. The right external jugular vein was separated. After blocking the distal vein using a hemostatic clip, we dissociated a 1.0-cm vein bridge and flushed the lumen with a physiological saline solution until it was completely dilated. We separated a 2.0-cm segment of the right common carotid artery, ligated the proximal and distal ends of the artery to block blood flow using vascular clamps, and opened a 1.0-cm incision near the arterial side. We connected the vein to the artery via an interrupted suture (11.0) using three square knots to ensure patency of the anastomosis. For the sham control group, the right submandibular gland and right sternocleidomastoid muscle were freed and resected as above, the right external jugular vein and right carotid artery were carefully separated, and the neck incision was sutured. We confirmed the absence of bleeding before suturing the neck incision, layer by layer. Rats were placed in cages and monitored continuously for 30 min until awake. Two groups of rats were studied after AVF surgery: a group treated with vehicle (dimethyl sulfoxide [DMSO], IP injection; n = 5 rats/group) and a group treated with Mdivi-1 (50 mg/kg in DMSO, IP injection; five rats/group) given 60 min before and 6 h after surgery. The vascular samples were harvested at 4 weeks after the surgery.
HE and Masson staining
Human and rat vascular samples used for protein extraction were rapidly washed and immersed in liquid nitrogen before being stored at −80°C. For immunohistochemical studies, vascular samples were fixed in fresh 4% paraformaldehyde, embedded in paraffin, and cut into 4-μm sections. The sections for HE and Masson staining were dewaxed, rehydrated, and stained with hematoxylin and eosin sequentially and transparently as described previously. 25 The intima/media (I/M) ratio was calculated by Image-Pro Plus 6.0 image analysis software.
Immunohistochemistry analysis
Sections were dewaxed and rehydrated, and then stained with antibodies for proliferating cell nuclear antigen (PCNA, 1:1000, ab29, Abcam, Cambridge, UK), alpha-smooth muscle actin (α-SMA, 1:1000, ab5694, Abcam), dynamin-related protein (Drp1, 1:5000, ab184247, Abcam), and phosphorylation of Drp1 at Ser-616 (p-Drp1 (616), 1:5000, 4494, Cell Signaling Technology, Danvers, MA, USA). Immunoreactivity was detected by sequential incubations of the vascular section with a biotinylated secondary antibody. Integrated optical density (IOD), calculated by IPP 6.0 software, was used to examine relative protein expression. The semi-quantitative analysis of the immunostained sections was performed by Image J software (National Institutes of Health, Bethesda, MD, USA). The human vascular tissues from 17 individuals of each group were prepared to determine the expression levels of target proteins. The average stained field was quantified from five randomly selected regions for each tissue section.
Cell culture of vascular smooth muscle cells
Primary VSMCs were prepared from male Sprague Dawley rats (250–300 g) as described. 26 Briefly, rats were euthanized by cervical dislocation under anesthesia with an intraperitoneal injection of sodium pentobarbital (50 mg/kg); VSMCs were isolated from the media of rat thoracic aortas. Aortas were cut into small pieces and allowed to attach to the plate. Explants were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 20% fetal bovine serum (FBS) in a 37°C incubator containing 5% CO2. Cells at passages 3–6 were used for all experiments.
Immunofluorescence
For staining of cultured cells, VSMCs were cultured using a chambered cover glass system (Thermo Scientific) for 2 days until they adhered to glass sides and were then treated with PDGF-BB (30 ng/mL) for 12 h. Cells were washed with PBS and fixed with 4% paraformaldehyde for 10 min. After three washes with PBS, cells were permeabilized with 0.2% Triton X-100 (Sigma) for 15 min and then washed with PBS. Cells were blocked with animal-free blocking solution (1x, Cell Signal Technology) for 30 min at room temperature and then anti-TOM20 antibody (1:200, 11802-1-AP, Proteintech, Rosemont, IL, USA) at 4°C overnight. After washing with PBS, cells were incubated with secondary antibodies for 2 h at room temperature. Slides were mounted with ProLong Gold mounting medium with DAPI (Molecular Probes) and covered with a coverslip. A fluorescence microscope system (Olympus, Japan) was used for imaging.
Vascular smooth muscle cell migration assays
Scratch-wound assays and modified transwell assays were performed as described. 26 For the scratch-wound assay, 1x106 VSMCs/mL were seeded onto gelatin-coated 6-cm dishes and allowed to form confluent monolayers. Cells were mechanically scraped from the bottom of the culture plate using a sterile pipette tip to create a cell-free (wounded) area and then washed with medium to remove debris. Wounded monolayers were incubated in medium with or without PDGF-BB (30 ng/mL) for 24 h. Rat VSMCs were pre-treated with a PI3K inhibitor (LY294002, 10 μM), an Akt1/2 kinase inhibitor (IH, 10 μM), or mitochondrial fission inhibitor (M, 10 μM) for 60 min. The average migration field was quantified on the 8 to 10 images of cells utilizing Image J software (National Institutes of Health, Bethesda, MD, USA). Assays were performed independently 5 times.
Modified transwell migration assays were performed in 24-well tissue culture plates. VSMCs (1 × 106 cells/mL) were seeded in the inner chamber of the transwell (polycarbonate membranes with 8-μm pores [Corning, NY, USA]), which were then placed into the outer chamber filled with 600 μL of serum-free DMEM containing PDGF-BB (30 ng/mL) and incubated at 37°C for 24 h. Rat VSMCs were pre-treated with a PI3K inhibitor (LY294002, 10 μM), an Akt1/2 kinase inhibitor (IH, 10 μM), or mitochondrial fission inhibitor (M, 10 μM) for 60 min. Cells that migrated to the outer surface of the membrane were fixed with 4% paraformaldehyde and were stained with DAPI. The number of migrated cells was counted in 4–6 randomly chosen fields of duplicate chambers at × 200 magnification for each sample. Assays were performed independently 5 times.
CCK8 assay
Vascular smooth muscle cell growth was assessed with the CCK8 assay kit (Dojindo, Japan), following the kit instructions. 27 Briefly, VSMCs were plated in 96-well plates, and treated with PDGF-BB (30 ng/mL, 24 h), Mdivi-1 (10 μM), PI3K inhibitor (LY294002, 10 μM), and an Akt1/2 kinase inhibitor (IH, 10 μM) for 60 min. After VSMCs were incubated with 10 μL of CCK8 solution for 30 min at 37°C, the absorbance of each well was measured at 450 nm using a spectrophotometer (Thermo Fisher Scientific, USA). VSMC viability was determined for the cells in each control group and compared with that of untreated VSMCs. Assays were performed independently 5 times.
RNA isolation and quantitative real-time PCR (qRT-PCR) analysis
Total RNA was extracted from rat vascular tissues using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) as previously described. 28 RNA was quantified by absorbance at 260 nm. Complementary DNA (cDNA) was then synthesized using the One-Step gDNA Removal and cDNA Synthesis SuperMix (TransGen, Beijing, China). qRT-PCR analysis was performed on an ABI-7500 Real-Time PCR System using Green qPCR SuperMix (TransGen, Beijing, China) reagent. The β-actin gene was used as an internal reference to evaluate the relative gene expression level. Relative expression of target genes was calculated using the 2−ΔΔCt method. 29 Assays were performed independently 5 times.
Western blot analysis
Vascular tissues or VSMCs were placed in a RIPA buffer with a protease inhibitor cocktail (1 m
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). Statistical differences were analyzed by independent sample t-test. Prism version 8.0 (GraphPad) was used for statistical analyses. Differences were considered significant when p < 0.05.
Results
Comparison of clinical data of the two groups of patients
There were no significant differences in gender, age, smoking history, diabetes, or hypertension between the two groups (Table 1).
Abnormal proliferation of VSMCs may affect AVF neo-intimal formation
Abnormal proliferation and migration of VSMCs can occur in injured vessels.30–32 However, there is no direct evidence that this alteration causes neo-intimal formation in AVFs. To address this knowledge gap, we examined longitudinal sections of human AVF stenosis tissues. Samples from the AVF group had significantly increased intima-media ratio (I/M) (Figure 1(a)) and levels of fibrosis (Figure 1(b)) compared to the control group. Levels of PCNA and MMP2 mRNA and protein also were increased in the AVF group compared to controls (Figures 1(c) and (d)). Immunohistochemistry was used to assess the expression of both PCNA and MMP2 (Figure 1(e)). PCNA and MMP2 were significantly higher in the AVF group compared to controls. Abnormal proliferation of VSMCs may affect AVF neo-intimal formation. (A) Representative HE staining and overall quantitation of intima/media (I/M) ratio in AVF and control patient groups (n = 5). (B) Representative Masson staining and overall quantitation of fibrosis in AVF and control groups (n = 5). Integrated optical density (IOD) as a measure of pixel intensity and area. (C) Western blots and corresponding quantitation for PCNA and MMP2 in vascular tissues from AVF and control groups (n = 3). β-actin is a loading control. (D) qRT-PCR for PCNA and MMP2 expression in vascular tissues from AVF and control groups (n = 5). (E) Immunohistochemistry staining showed higher expression of PCNA and MMP2 in AVF. **p < 0.01, ****p < 0.0001.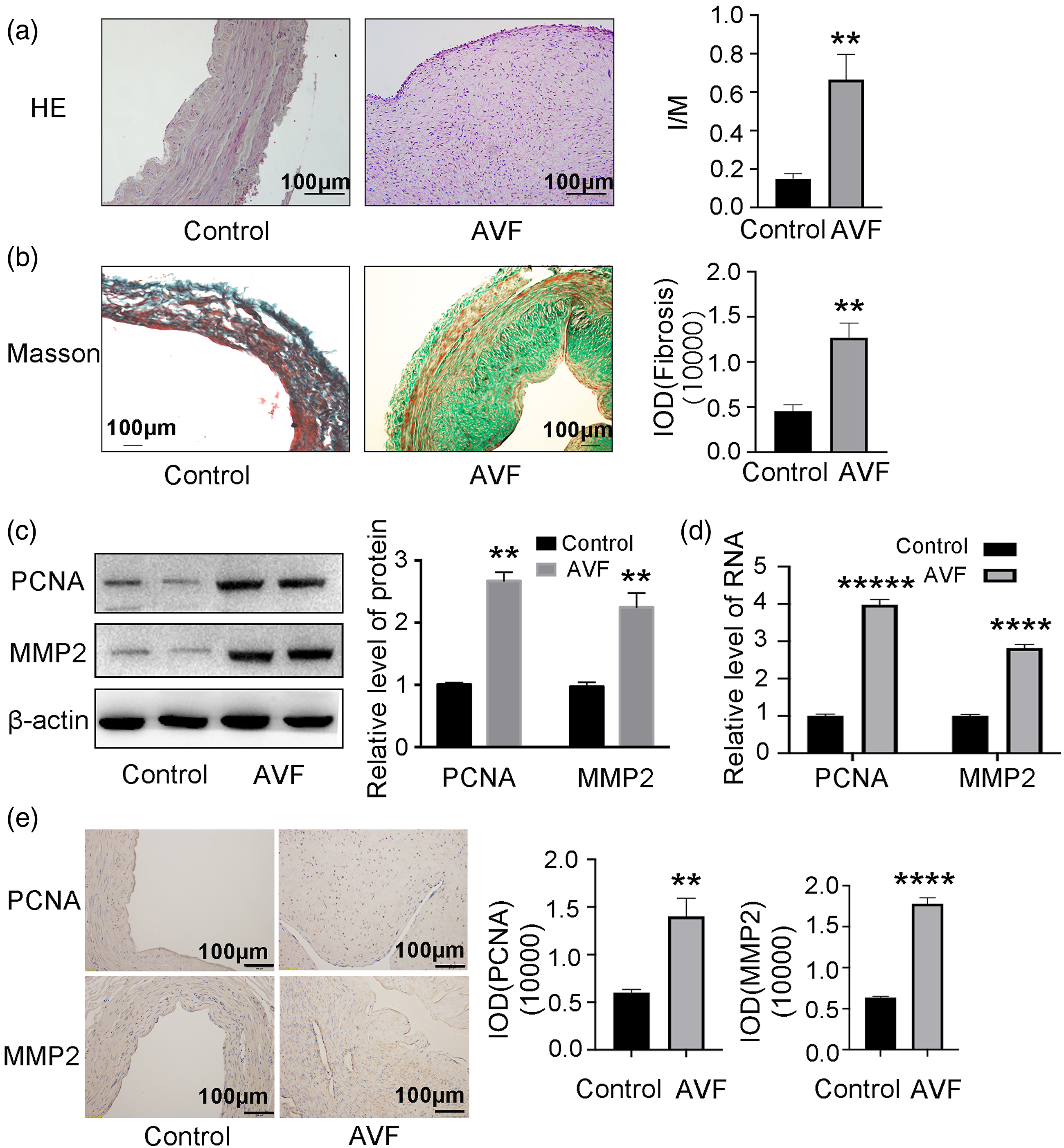
PGDF-BB signaling affects VSMC proliferation/migration via a mechanism involving mitochondrial fission
PDGF is reported to be a vascular remodeling activator,33,34 so we investigated PDGF-BB expression in human AVF stenosis and control veins. Protein levels of PDGF-BB were significantly higher in the AVF group compared to controls (Figure 2(a)). To test the effect of PDGF-BB in the pathophysiology of VSMC responses, we assessed proliferation and migration of cultured rat VSMCs. PDGF-BB significantly stimulated VSMC proliferation in a CCK8 assay (Figure 2(b)) and migration in the scratch wound (Figure 2(c)) and transwell assays (Figure 2(d)) compared to controls, suggesting that PDGF-BB may contribute to neo-intimal formation by promoting VSMC proliferation/migration. PGDF signaling affects VSMC proliferation/migration via a mechanism involving mitochondrial fission. (A) Western blots and corresponding quantitation of protein levels of PDGF-BB in human AVF stenosis and control veins (n = 3). β-actin is a loading control. (B) PDGF-BB stimulated rat VSMC proliferation, and Mdivi-1 (M) blunted this effect in a CCK8 assay at days 1, 2, 3, and 4 (n = 5). PDGF-BB stimulated the rat VSMCs migration by (C) scratch wound and (D) transwell assay. Mdivi-1 blunted this effect at 24 h (n = 5). (E) Immunostaining for p-Drp1 (616) and corresponding quantitation in sections of veins from AVF and control patients (n = 5). (F) Western blots and corresponding quantitation of protein levels of Drp1, p-Drp1 (616), and PCNA in rat VSMCs after treatment with PDGF-BB or PDGF-BB + M (n = 5). β-actin is a loading control. (G) Representative confocal images of rat VSMCs stained for the mitochondrial marker TOM20 after treatment with PDGF-BB or PDGF-BB + M. Control set as 1 for relative fold change. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 VS control group. #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 VS PDGF-BB group.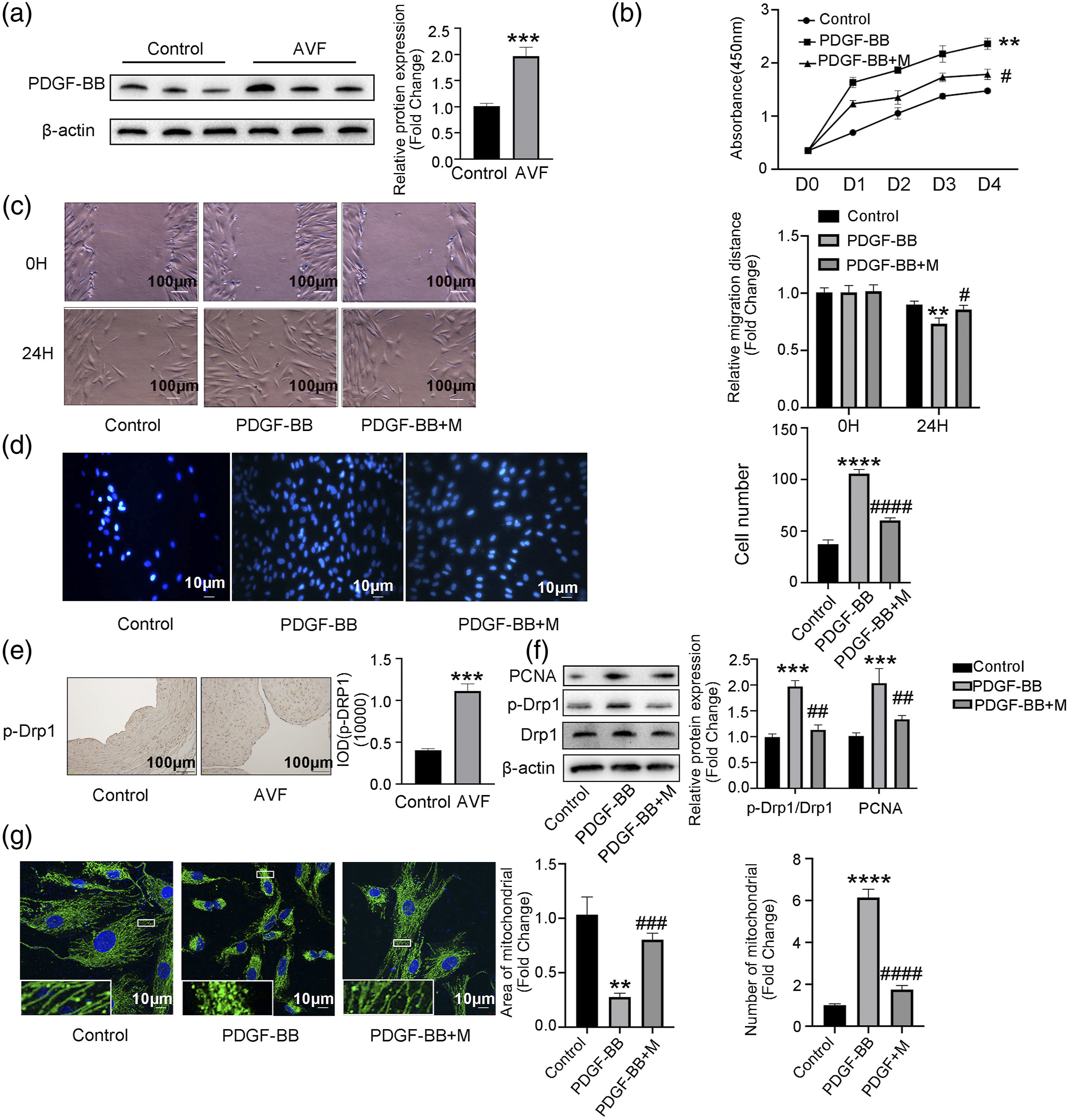
Recent studies demonstrated that mitochondrial fission is involved in vascular remodeling,14,35 but it is unclear if mitochondrial fission contributes to AVF neo-intimal formation. We found that human AVF tissues have elevated expression of p-Drp1 (616) (Figure 2(e)) and that PDGF-BB treatment increases expression of p-Drp1 (616) in cultured rat VSMCs (Figure 2(f)). We also examined mitochondria in rat VSMCs using confocal microscopy and found that PDGF-BB treatment reduced mitochondrial size and length in VSMCs (Figure 2(g)), consistent with p-Drp1 (616) expression results.
To test if mitochondrial fission acts downstream of PDGF-BB to control VSMC behaviors, we assessed cells treated with the fission inhibitor Mdivi-1 (M). Importantly, Mdivi-1 (10 μM, 60 min) blunted the effects of PDGF-BB on VSMC proliferation and migration (Figure 2(b)–(d)). Mdivi-1 treatment also blocked the PDGF-BB–mediated increase in p-Drp1 (616) expression (Figures 2(e) and (f)) and the PDGF-BB–mediated decrease in mitochondrial size (Figure 2(g)).
PDGF-BB affects VSMC proliferation/migration and mitochondrial fission via the PI3K/Akt pathway
PDGF signaling plays an active role in the PI3K/Akt signaling pathway.26,36 Further, the overexpression of PDGF in endothelial progenitor cells promotes vascular repair in the early phase after vascular injury through enhanced cell proliferation, migration, and angiogenesis.37,38 However, the relationship between PI3K/Akt signaling and cell proliferation/migration and mitochondrial dynamics in VSMC is unknown. We found that phosphorylation of Akt at Ser-473 was elevated in human AVF tissue (Figure 3(a)) and that pretreatment of rat VSMCs with a PI3K inhibitor (LY294002, 10 μM) or an Akt1/2 kinase inhibitor (IH, 10 μM) for 60 min blocked PDGF-BB–induced VSMC proliferation/migration (assays: scratch wound, Figure 3(b); transwell, Figure 3(c); and CCK8, Figure 3(d)). Western blots and corresponding quantitation of protein levels of p-Drp1 (616) in rat VSMCs after treatment with PDGF-BB or PDGF-BB + LY294002 or PDGF-BB + IH. Additionally, inhibition of PI3K and Akt activation also corrected the excessive mitochondrial fission and phosphorylation of Drp1 at Ser-616 induced by PDGF-BB (Figure 3(e)). We examined mitochondria in rat VSMCs using confocal microscopy and found that LY294002 or IH treatment increased mitochondrial size and length in VSMCs induced by PDGF-BB (Figure 2(f)). PDGF-BB affects VSMC proliferation/migration and mitochondrial fission via the PI3K/Akt pathway. (A) Western blots and corresponding quantitation of protein levels of Akt and p-Akt in human AVF stenosis vessels and control veins (n = 3). (B) Scratch-wound assay, (C) transwell assay, and (D) CCK8 assay of rat VSMCs pre-incubated with the PI3K inhibitor LY294002 (LY, 10 μM) or Akt1/2 kinase inhibitor (IH, 10 μM) for 60 min and then treated with PDGF-BB (n = 5) for 24 h. (E) Western blots and corresponding quantitation of protein levels of Drp1 and p-Drp1 (616) from VSMCs treated with PDGF-BB, PDGF-BB + LY, or PDGF-BB + IH (n = 3). (F) Immunostaining of VSMCs for the mitochondrial marker TOM20 after treatment with PDGF-BB, PDGF-BB + LY, or PDGF-BB + IH. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 VS PDGF-BB group.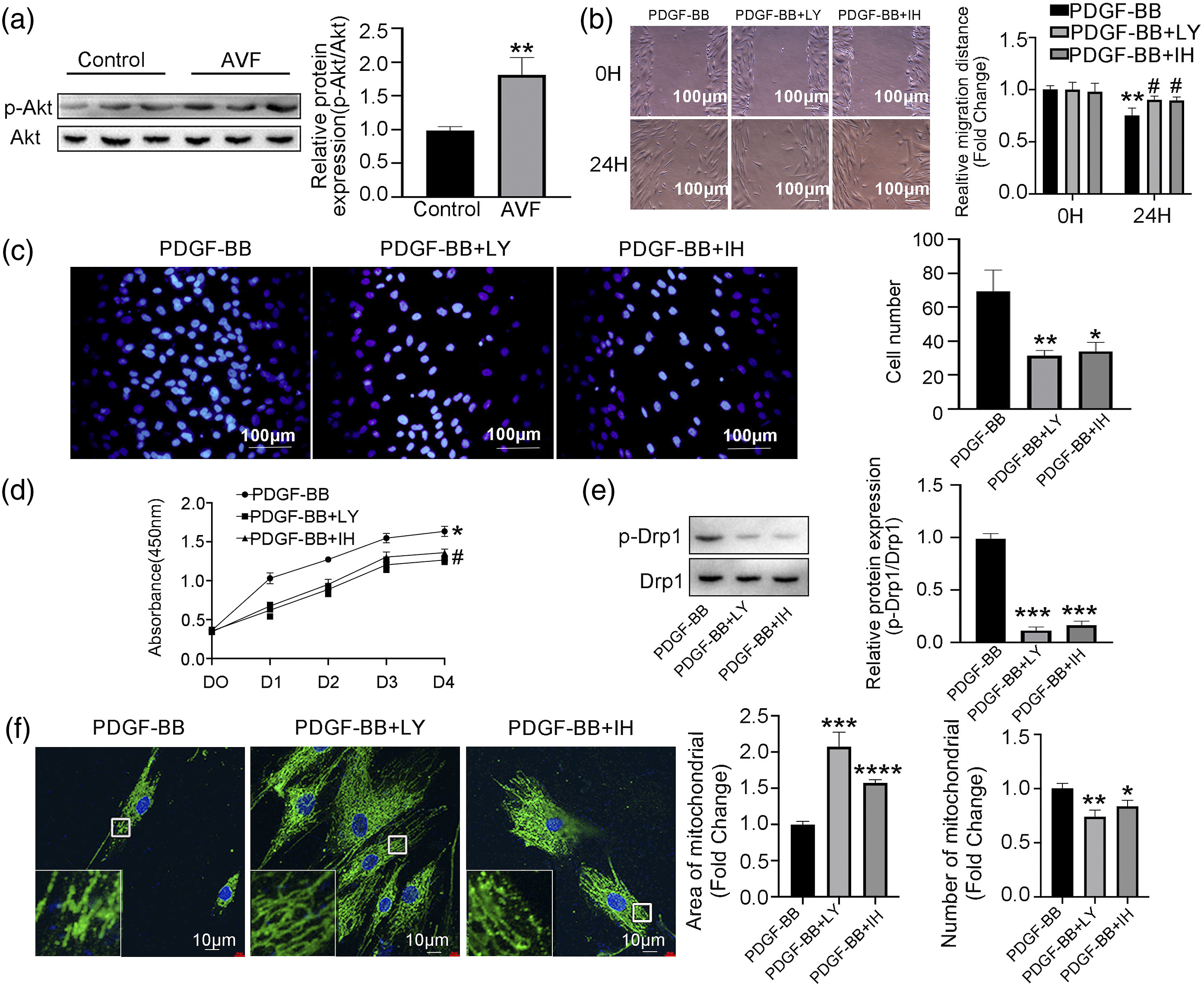
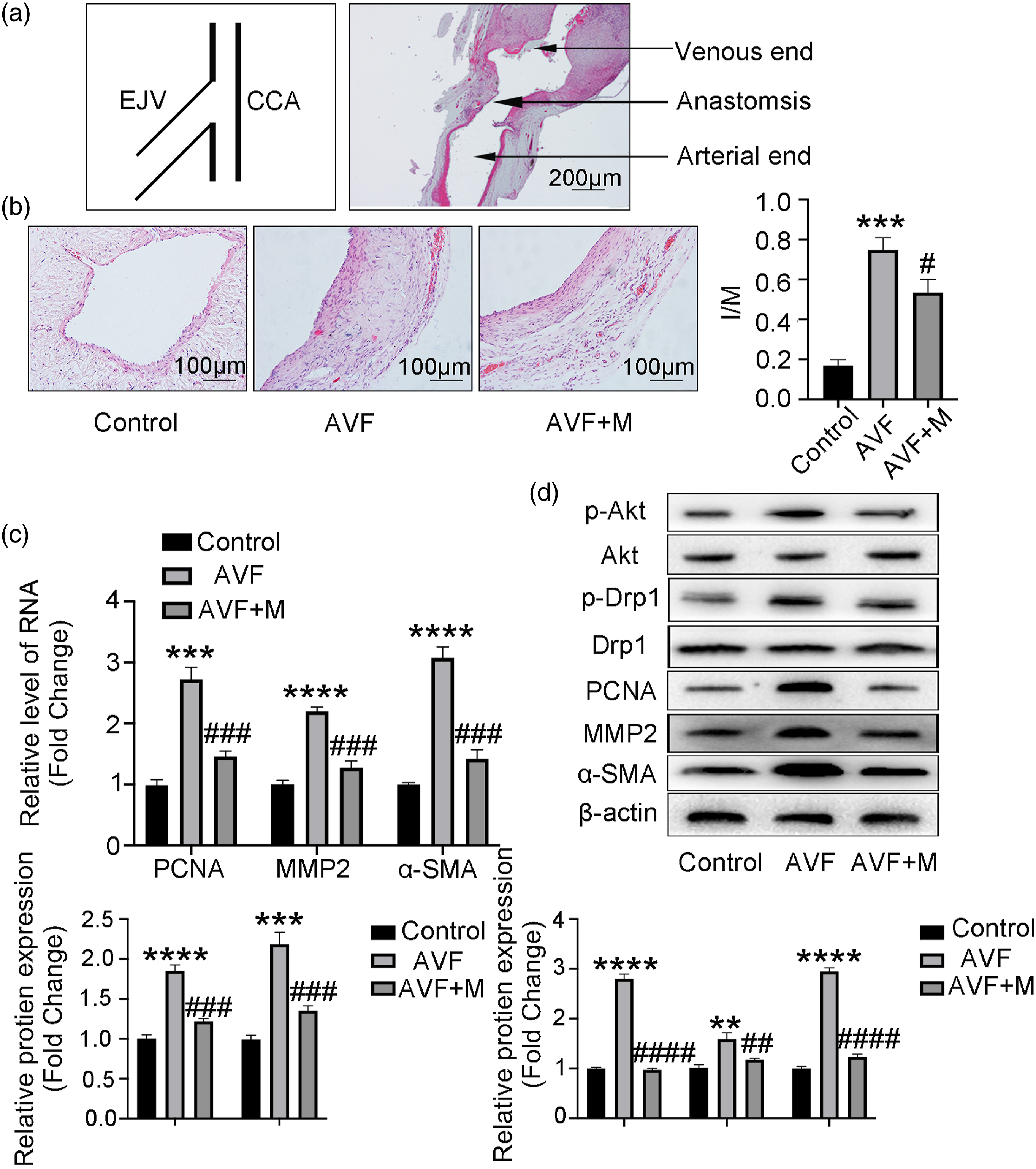
Inhibition of mitochondrial fission attenuates neo-intimal formation in a rat AVF model
To directly test whether inhibition of mitochondrial fission is sufficient to attenuate neo-intimal formation, we used a rat AVF model (Figure 4). Rats were subjected to intraperitoneal injection with mitochondrial fission inhibitor Mdivi-1 after development of AVF. The schema and representative histology of a common carotid artery/external jugular vein anastomosis is shown in Figure 5(a). HE staining revealed a significant increase in intima-media ratio (I/M) in the AVF group compared to controls 2 weeks after establishing the rat model, and Mdivi-1 treatment nearly abrogated this effect, suggesting an essential role of mitochondrial fission in neo-intimal formation (Figure 5(b)). We also evaluated VSMC proliferation by PCR (Figure 5(c)) after establishing the rat model and detected the expression of p-Akt, p-Drp1, PCNA, MMP2, and α-SMA by Western blot (Figure 5(d)). We found that inhibition of mitochondrial fission could reverse VSMC proliferation and attenuate neo-intimal formation in a rat AVF model via the PI3K/Akt pathway. Surgical procedure for creating a rat AVF model. (A) Positioning of the animal. (B) Rats were anesthetized with 75 mg/kg of pentobarbital, and the neck skin was disinfected with iodophor. (C) A 1.5-cm oblique incision was made in the neck, slightly off-center. (D, E) The right submandibular gland and right sternocleidomastoid muscle were freed and resected. (F) We separated a 2.0-cm segment of the right common carotid artery, and ligated the proximal and distal ends of the artery to block blood flow using vascular clamps. (G, H) The right external jugular vein was separated. (I, J) After blocking the distal vein using a hemostatic clip, we dissociated a 1.0-cm vein bridge and flushed the lumen with a physiological saline solution until it was completely dilated. (K) We opened a 1.0-cm incision near the arterial side. (L) We connected the vein to the artery via an interrupted suture (11.0) using three square knots to ensure patency of the anastomosis. Inhibition of mitochondrial fission attenuates neo-intimal formation in a rat AVF model. (A) Schema of the anastomosis and representative histology. EJV: external jugular vein; CCA: common carotid artery. (B) Representative HE staining and quantitation of intima/media (I/M) ratios of control, AVF, and Mdivi-1 (M)-treated AVF groups (n = 5). (C) qRT-PCR for PCNA, MMP2, and α-SMA expression in vascular tissues from control, AVF, and AVF + M groups (n = 5). (D) Western blots and corresponding quantitation of p-Drp1 (616), p-Akt, PCNA, MMP2, and α-SMA in control, AVF, and AVF + M groups (n = 5). β-Actin is a loading control. *p < 0 .05, **p < 0.01, ***p < 0.001, ****p < 0.0001 VS control group. #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 VS AVF group.
Discussion
AVF primarily fail because of three issues: early thrombosis, a failure to mature due to aggressive neo-intima hyperplasia or impaired outward remodeling, or the effect of the anastomotic configuration and repeated needle puncturing. 23 While VSMC proliferation and migration reportedly play a role in the pathogenesis of failed dialysis accesses,10,39 the underlying mechanisms have remained unclear. The extensive adverse consequences resulting from AVF failure exerts a burden on hemodialysis patients highlights the need for pathophysiological studies to unravel the complex mechanisms underlying AVF failure. Here, we demonstrated two linked cellular events: mitochondrial fission and abnormal proliferation and migration of VSMCs in neo-intima formation during AVF failure. We observed that inhibition of mitochondrial fission could decrease proliferation/migration of VSMCs and inhibit neo-intimal formation in vivo and in vitro. In addition, these effects were associated with PI3K/Akt activation. We believe that these results are the first evidence to demonstrate the relationship between mitochondrial fission and AVF neo-intimal formation. These findings provide new insights into mitochondria as a novel target with significant therapeutic potential for AVF patients.
We found that AVF stenosis was associated with intimal hyperplasia, fibrosis, and VSMC proliferation. Such changes are likely integrated with the cells’ energetic state, suggesting mitochondrial regulation in VSMCs may play an important role in AVF stenosis. Mitochondrial fragmentation increased the mtROS levels, and decreased the ATP concentration in vascular endothelial cells 40 and lung microvascular endothelial cell. 41 Meanwhile, abnormal mitochondrial fission plays a crucial role in regulating inflammasome activation in retinal capillary endothelial cells. 42 The mitochondria in TGF-β1-treated cardiac fibroblasts were noticeably more fragmented than those of controls. 43 Moreover, excessive mitochondrial fission acts as a pro-proliferative marker in renal fibroblast activation in humans and mice. 44 Similarly, changes in mitochondrial dynamics are implicated in the development and progression of cardiovascular disease (CVD).14,22,35,45 Mounting evidence indicates that mitochondrial fission accelerates VSMC proliferation after vascular injury.13-15
To explore the relationship between mitochondrial regulation and abnormal VSMC proliferation/migration, we performed quantitative controlled culture and in vivo studies. We demonstrated that mitochondrial fission was markedly increased in tissue from AVF stenosis patients. Also, the level of a mitochondrial fission-related protein was significantly increased in cultured rat VSMCs treated with PDGF-BB. Importantly, an inhibitor of mitochondrial fission blocked the effect of PDGF-BB on VSMC migration/proliferation in culture and partially blocked neo-intimal formation in vivo. This strongly suggests that increased mitochondrial fission contributes to the VSMC hyper-proliferation and migration seen in vivo and vitro. The emerging role of mitochondrial regulation in AVF requires further studies to better elucidate the mechanisms controlling this process as well as its importance in AVF pathology.
Mechanistically, we identified PDGF-BB signaling and the Akt/PI3K pathway as important regulators of mitochondrial dynamics and the proliferation/migration of VSMCs. Activation of PI3K plays an important role in promoting cell survival and proliferation in numerous cell systems.46,47 Activation of PI3K results in recruitment of the serine-threonine kinase Akt, one of the downstream targets of PI3K, to the plasma membrane. Activated Akt then affects the activity or abundance of several transcription factors linked to cell survival and proliferation. In VSMCs, most growth factor-induced responses are mediated by PI3K/Akt signaling. 26 We found that PDGF-BB could induce Akt phosphorylation in cultured VSMCs and that PI3K inhibitor and Akt1/2 kinase inhibitor could block the effect of PDGF-BB on VSMC migration. We also found increased p-Akt expression in human AVF stenosis patients, indicating this pathway may play an important role in the development of AVF in humans. These signaling events are also linked to mitochondrial dynamics, as we found that PI3K inhibitor and Akt1/2 kinase inhibitor could diminish the mitochondrial fission induced by PDGF-BB.
In conclusion, we demonstrated that excessive mitochondrial fission promotes VSMC proliferation/migration by activation of PI3K/Akt and inhibition of mitochondrial fission could attenuate AVF neo-intimal formation. These findings improve our understanding of the molecular mechanisms involved in AVF neo-intimal formation and may provide new insights for future therapeutic targets for AVF stenosis patients (Figure 6). Graphic representation of major pathways implicated in mitochondrial fission based on the PDGF-BB effect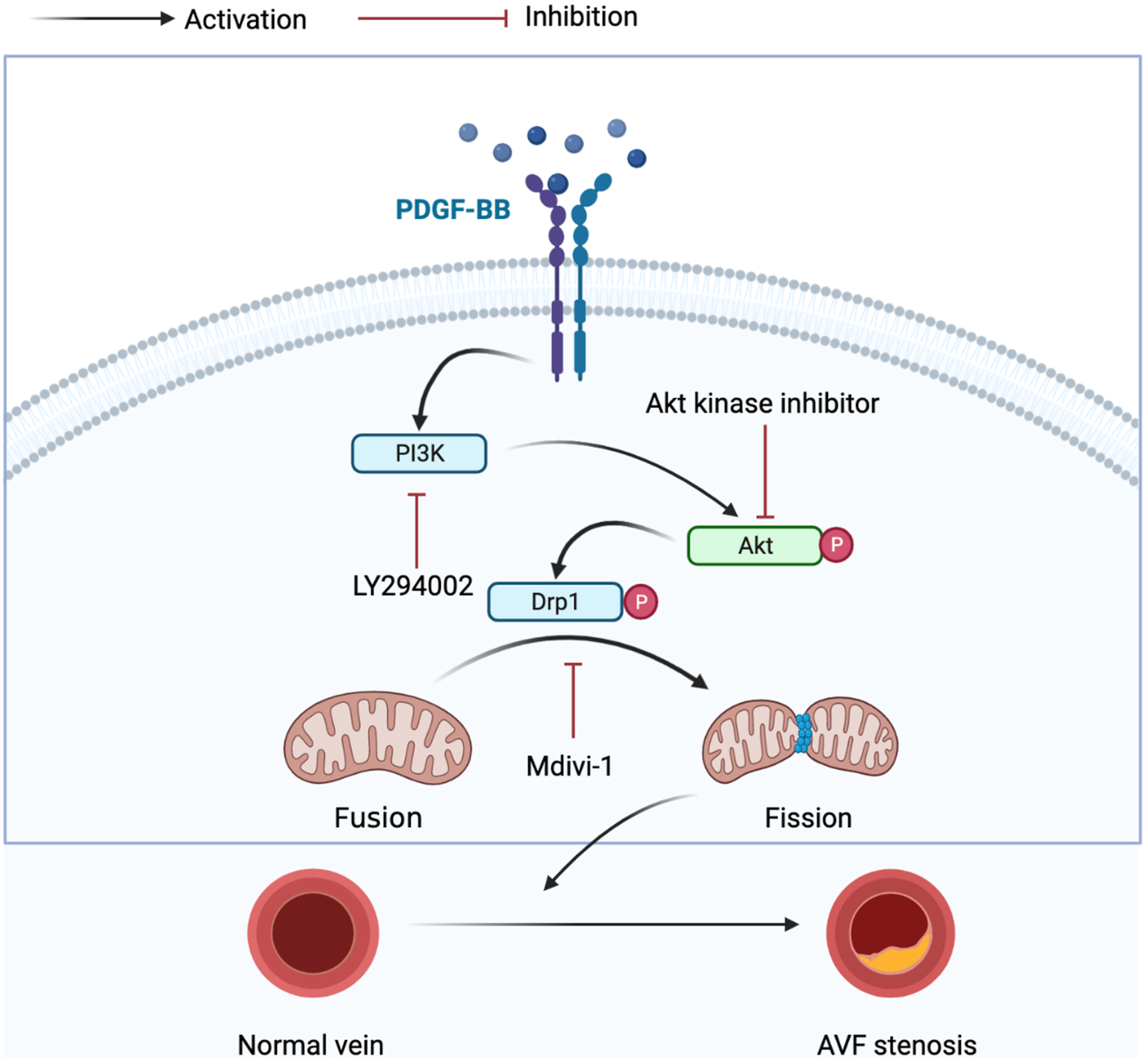
Because it would not be feasible to get a normal AVF sample to compare with, we could only choose vein specimens derived from patients undergoing coronary artery bypass surgery using the greater saphenous vein as a control. This may be a limitation in our study.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (81670443, 81670275) and the International S&T Cooperation Program (2013DFA31900).