
Abstract
This research discusses the role of new formulation strategies on life cycle management in the US pharmaceutical industry and evaluates its effect on the market life of existing drug products. We constructed a method to quantitatively measure the life cycle extension period of new formulation pharmaceuticals, including censored cases, wherein the generic drug has not yet been approved and the innovator drug is surviving in the market. We employed 180 new formulation approvals for which Abbreviated New Drug Application approvals were granted for new molecular entities. We undertook a comprehensive and quantitative analysis of how long these new formulation pharmaceuticals are able to prevent the entry of generic pharmaceuticals into the US market after the end of the regulatory exclusivity period. The results showed that among the oral formulations, dosage forms that require advanced manufacturing technology, such as “Extended/Delayed Release,” have significantly extended the life cycle extension period over the past 20 years. In addition, the acquisition of formulation-related patents protecting products was observed to extend the life cycle extension period significantly. These research outcomes can help business strategists in the pharmaceutical industry to take advantage of already marketed new molecular entities as part of the life cycle management strategy and can help generic drug companies adapt to the strategies of innovator drug companies.
The promotion of the medical expense restraint policy and the decreased efficiency of R&D in recent years have created an increasingly harsh business environment for innovator pharmaceutical companies around the world. In the US pharmaceutical market, the introduction of the Hatch–Waxman Act in 1984 encouraged the market entry of generic pharmaceutical companies. As a result, generic drugs have enjoyed a quantity share of more than 60% as of 2006.1–3 Life cycle management (LCM) that maximizes the value of existing drugs and extends product life is therefore a vital business strategy for innovator pharmaceutical companies. The LCM strategies of these companies include various techniques such as new indication (NI), new formulation (NF), new combination (NC), authorized generic, over-the-counter (OTC) drug switch, and pricing strategy.4–7 This study focuses on the NF strategy from among the various choices of LCM. NFs have accounted for the highest number of new drug application (NDA) approvals by the US Food and Drug Administration (FDA) in recent years (Figure 1). Whereas NME approval numbers peaked in the 1990s and then declined steeply, the number of NF approvals remained high even in the 2000s. These statistics underscore the heightening importance of the NF strategy, as NME creation has become increasingly difficult in recent years.
Timestamp indicating the number of FDA approvals for NF, NME, NC, NI, and OTC drug switch.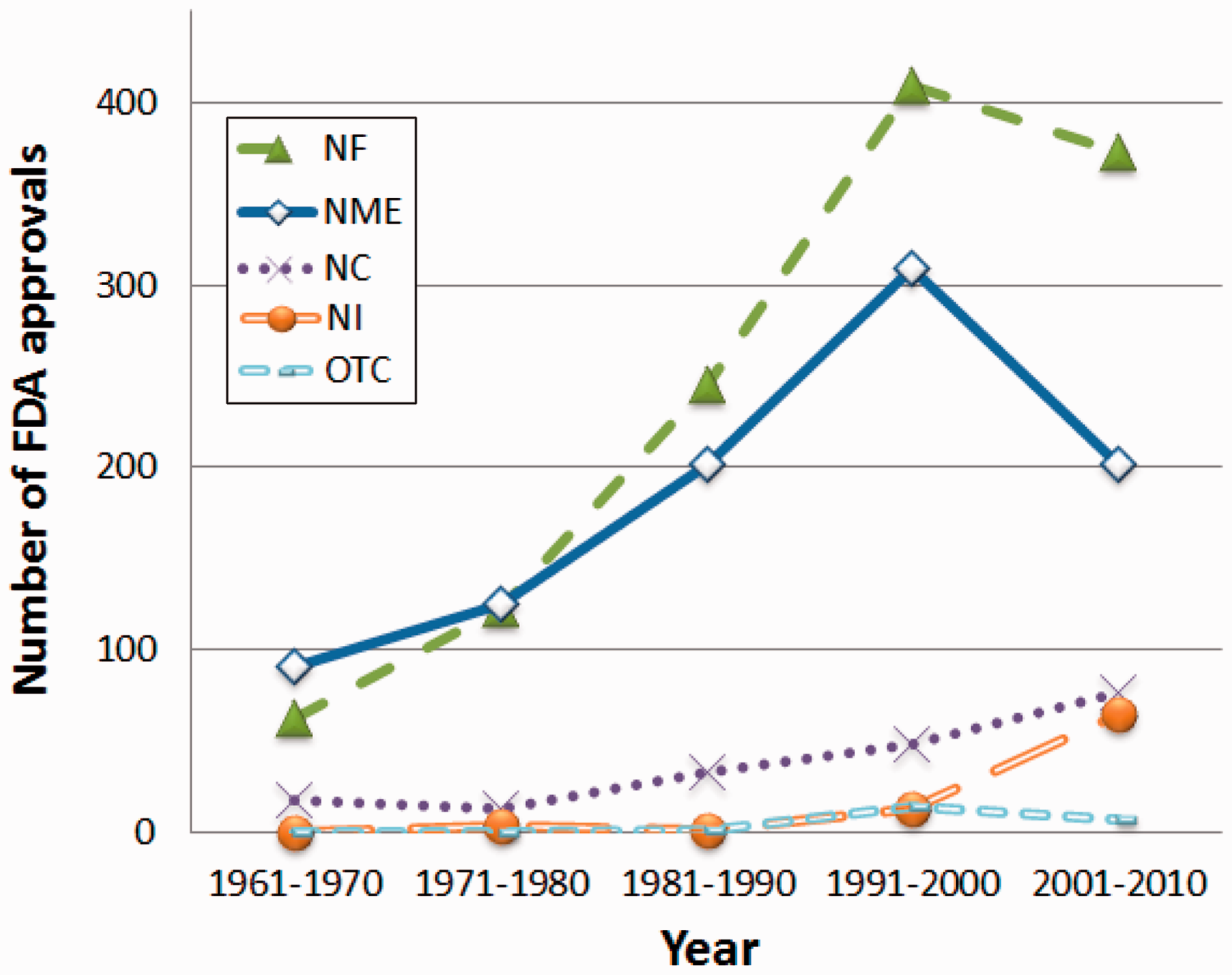
The NF strategy is intended to increase sales and extend the product life of an existing drug by improving its formulation. A detailed analysis of the NF strategy of innovator pharmaceutical companies is important to the efficient development and marketing of generic drugs by generic pharmaceutical companies.
In this study, we construct a quantitative analysis method capable of evaluating the innovator drug’s life cycle extension effects of NF strategies, in order to determine which option is the most effective from among the various NF types.
Background
There are numerous LCM case studies in the US pharmaceutical industry. Dubey and Dubey 8 examined case studies of NFs, NIs, and NCs—the three most commonly utilized LCM strategies—and suggested that it is possible to efficiently prevent generic drug entries by selecting a suitable LCM strategy.
Several studies have used statistical methods to comprehensively and quantitatively analyze various factors that affect LCM.9–12 Grabowski and Kyle 9 investigated NMEs for which generic drugs had entered the market within a certain time period and employed regression analysis taking the pharmaceutical exclusivity period as the explained variable to investigate the effects of pharmaceutical sales, Paragraph IV certification and chemical type (NMEs and NFs). Hemphill et al.10,11 used regression analysis to investigate the relationships between the effective exclusivity period and Paragraph IV certification, with various factors such as pharmaceutical sales. Morton 12 used statistical methods to conclude that the chronic disease sector and oral drugs attract generic pharmaceutical companies and that advertising is not a barrier to generic drug entry.
Among the studies focusing on NFs, Tony and Hansen 13 qualitatively explained that NFs had been the most commonly employed of all LCM strategies over the past 20 years and that one of the important roles of this strategy was to prevent the market entry of generic drugs. Manso and Sokol 14 gave several examples to show that the acquisition of additional approvals for various dosage forms and the sequential marketing of NFs successfully maintained the number of innovator drug prescriptions, even after generic drugs had entered the market. Dubey and Dubey 8 cited the examples of clarithromycin and bupropion, wherein the selection of suitable dosage forms meant that the innovator drugs were able to maintain a high market share even after generic drugs had entered the market. As a characteristic of the NF strategy, Dubey and Dubey also stated that while general formulations, such as tablets and capsules, are preferable development options (as these formulations pose few technical obstacles and offer short development times), they are also prone to rapid genericization. Dubey and Dubey 8 explained that in contrast to general formulations, dosage forms such as controlled-release formulations, which can be administered less frequently, are one of the most preferred formulations. Because such formulations require complex technology and are protected by patents, they could pose a greater barrier to the entry of generic drugs.
Certain studies have investigated the effects of patents on generic drug entry.15–17 Howard15,16 conducted a case study to evaluate the effect of the active substance patent strategy on the exclusivity period of fluvastatin and atorvastatin. They stated that only substance patents can extend the exclusivity periods of pharmaceuticals, unless secondary patents suppress the development of circumvention technologies. Kapczynski et al. 17 performed a comprehensive analysis of secondary patents listed in the Orange Book and compiled a list of formulation patents. They found that formulation patents added an average of 6.5 years to the nominal exclusivity period of a drug after the expiry of the substance patent.
Most prior case studies have been more qualitative in nature. A few studies have discussed LCM strategies comprehensively and quantitatively, and even fewer studies have analyzed the NF strategy comprehensively and empirically. As stated earlier, according to Dubey and Dubey, 8 controlled-release formulations require a complex formulation technology and thus pose a greater barrier to the entry of generic drugs than general formulations such as tablets and capsules. However, there are hardly any quantitative investigations concerning the extent to which the dosage forms selected by innovator pharmaceutical companies deter generic drug entry.
Furthermore, with regard to almost all quantitative studies, regression analysis can only be applied to cases in which generic drugs have already entered the market. For example, it does not consider so-called “censored cases,” wherein the generic drug has not yet been approved and the innovator drug is surviving in the market. It also does not consider cases wherein the innovator drug is withdrawn from the market before generic drug entry. We believe it is important to include such “censored cases” in our analysis in order to improve accuracy and enable a comprehensive quantitative investigation. Therefore, this study employs the survival analysis method.
The pharmaceutical exclusivity period in the United States
We provide an overview of the unique pharmaceutical regulatory system in the United States.18–24
Abbreviated New Drug Application
If it can be demonstrated that the generic drug is bioequivalent to its respective innovator drugs, the generic pharmaceutical company can manufacture and market the generic pharmaceutical without repeated clinical trials and high development costs. However, this is only possible on the condition that the generic pharmaceutical has the same profile of the innovator drug in terms of active compound, dosage form, content, route of administration, etc.
Orange Book and Paragraph IV
As for patent exclusivity, a term of 20 years from the patent application filing date is generally granted. In the United States, pharmaceutical companies are also obliged to disclose information regarding patents that protect pharmaceuticals. This disclosed patent information is compiled in the Orange Book. During the time these patents appear in the Orange Book, any generic pharmaceutical company filing an Abbreviated New Drug Application (ANDA) must assert that it will not infringe the patent (Paragraph IV certification). Furthermore, because the FDA will suspend ANDA approval for the period of time the patent holder files a suit, the listing of patents in the Orange Book is a crucial procedure of the LCM strategy for innovator pharmaceutical companies.
Regulatory exclusivity
There are six types of regulatory exclusivity (RE) in the United States. The exclusivity periods for an NME, an NF, an NI, an orphan drug (OD), a pediatric clinical trial (PED), and the first ANDA filer with a Paragraph IV certification are 5 years, 3 years, 3 years, 7 years, 6 months, and 180 days, respectively. Market entry by generic pharmaceutical companies is generally not permitted during these exclusivity periods.
Materials and methods
Excluding approvals for combination drugs, there were 301 NF approvals with the NME and the NF from the same sponsor from January 1991 to December 2010. Of these 301 approvals, we use the 180 NF approvals for which ANDA approvals were granted for the NMEs, to analyze the pharmaceutical life cycle extension period (LEP).
Information regarding approvals for innovator and generic pharmaceuticals was acquired from the FDA website “Drugs@FDA.” 25 Information regarding innovator drug patents and regulatory exclusivity was acquired from the Orange Book for the years 1995 to 2011. Formulation-related patents were identified based on the Orange Book by deleting those patents that protect NME products from the list of patents that protect NF products.
The LEP owing to NF approvals was calculated starting from the date of the ANDA approval for the NME and defined as the period of time taken until ANDA approval is granted for the NF (Figure 2). In addition, as generic pharmaceuticals are denied market entry during the RE period, this period was subtracted from the LEP. Although the RE period for NFs is generally 3 years, in case RE for an OD approval was granted at the same time as that for an NF approval, we considered the RE period to be 7 years instead of 3 years.
Methodology for the quantitative measurement of the LEP of NF pharmaceuticals. REs of 5 years and 3 years are recognized for an NME and an NF.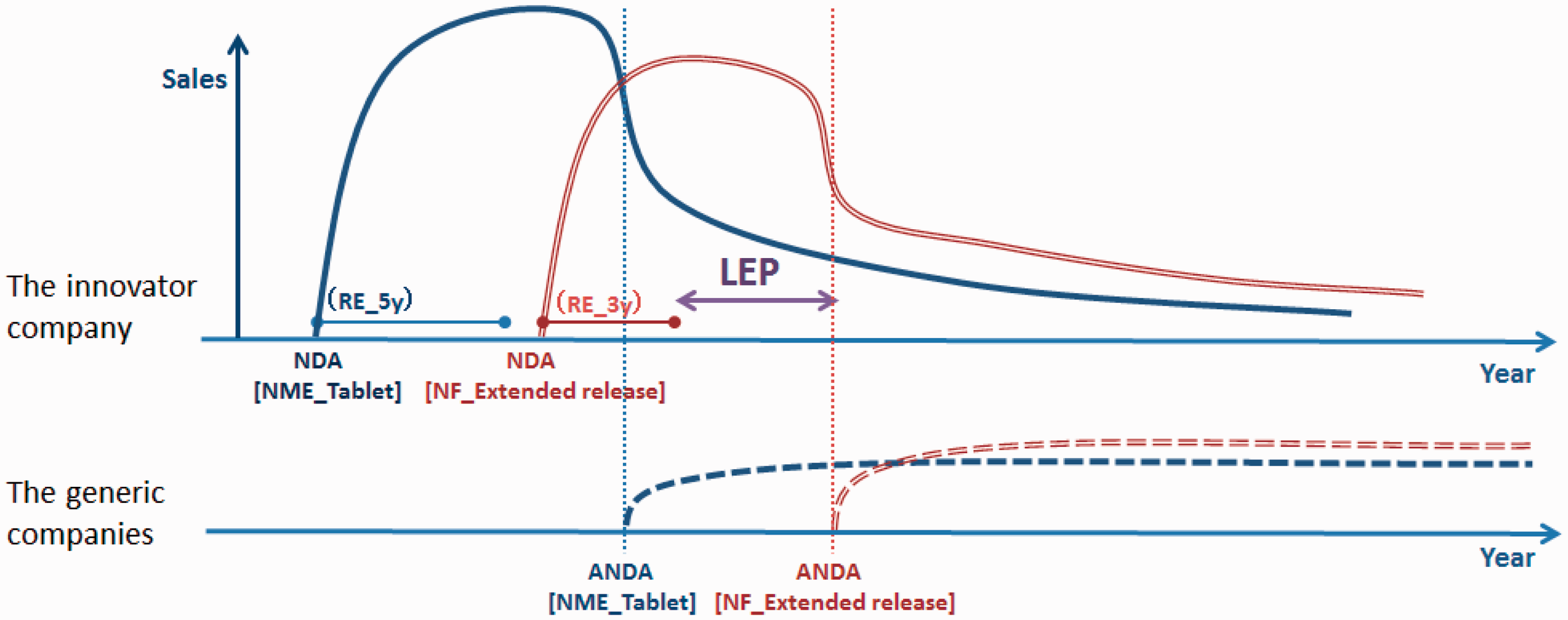
For the purpose of survival analysis, the formulation categories defined by the FDA were regrouped by route of administration into the categories “Oral” and “Non-Oral.” The latter included formulations such as “Injection” and “Ophthalmic.” The “Oral” category included “Oral-General Formulation” comprising general formulations such as tablets and capsules, “Extended/Delayed Release” comprising delayed release and extended release formulations and “Other Oral” comprising suspensions, solutions, syrups and so on. In this study, survival analysis (Kaplan–Meier method) was applied to a total of 180 NF approvals, comprising 96 approvals for which ANDA approvals were granted for both the NME and the NF and 84 approvals for which ANDA approval was granted for the NME only. Importantly, the regression analysis is not applicable to these 84 approvals, because it is not possible to handle “censored cases,” as described above. However, 7 of these 180 approvals are exceptional cases, in which the NF ANDA had been approved before the NME ANDA. These cases are defined as “day 0 LEP,” because it is regarded that they did not fulfill the intended role of the NF pharmaceuticals, namely to block generic drug entry. The starting date of the survival analysis is determined by calculating the starting point as described above, and the date of the ANDA approval for the NF serves as the endpoint. The observation period ranged from January 1990 to December 2011. Cases wherein the generic drug was not approved in NF pharmaceuticals during the observation period or the innovator drug was withdrawn from the market were considered to be censored cases. As a consequence, we plotted the Kaplan–Meier graphs for each LEP and employed the log-rank or Wilcoxon test to determine whether these delays differ significantly based on various conditions. In the survival analysis, the log-rank test gives more weight to later events, and the Wilcoxon test, to earlier events. The survival analysis of formulation-related patents employs data on patents listed in the Orange Book published 3 years after NF approval. Chronological changes in patent numbers during the observation period were not considered and were treated as being constant.
Results
First, we investigate the NF strategies of innovator pharmaceutical companies over the last 20 years. We utilize the dosage form categories and administration routes defined by the FDA
26
to determine the proportions of different dosage forms used for NME and NF pharmaceuticals from 1991 to 2010. The findings are summarized in Figure 3(a) and (b). For NME pharmaceuticals, approximately 70% of approvals were granted for the dosage forms “Oral-General Formulation” (comprising formulations such as tablets and capsules that do not require any special formulation technology) and “Injection” (Figure 3(a)). In contrast, for NF pharmaceuticals, the top three dosage forms accounted for only about 29% of all approvals, but the proportion of each formulation other than those in the top three is less than 5%. In other words, more than 70% of the total comprises formulations other than those in the top three (Figure 3(b)). The trend that more diverse dosage forms were selected for NF pharmaceuticals rather than NME pharmaceuticals was confirmed.
(a) Percentage of the top seven NME dosage forms and administration routes of 511 FDA approvals (1991–2010). (b) Percentage of the top seven NF dosage forms and administration routes of 781 FDA approvals (1991–2010).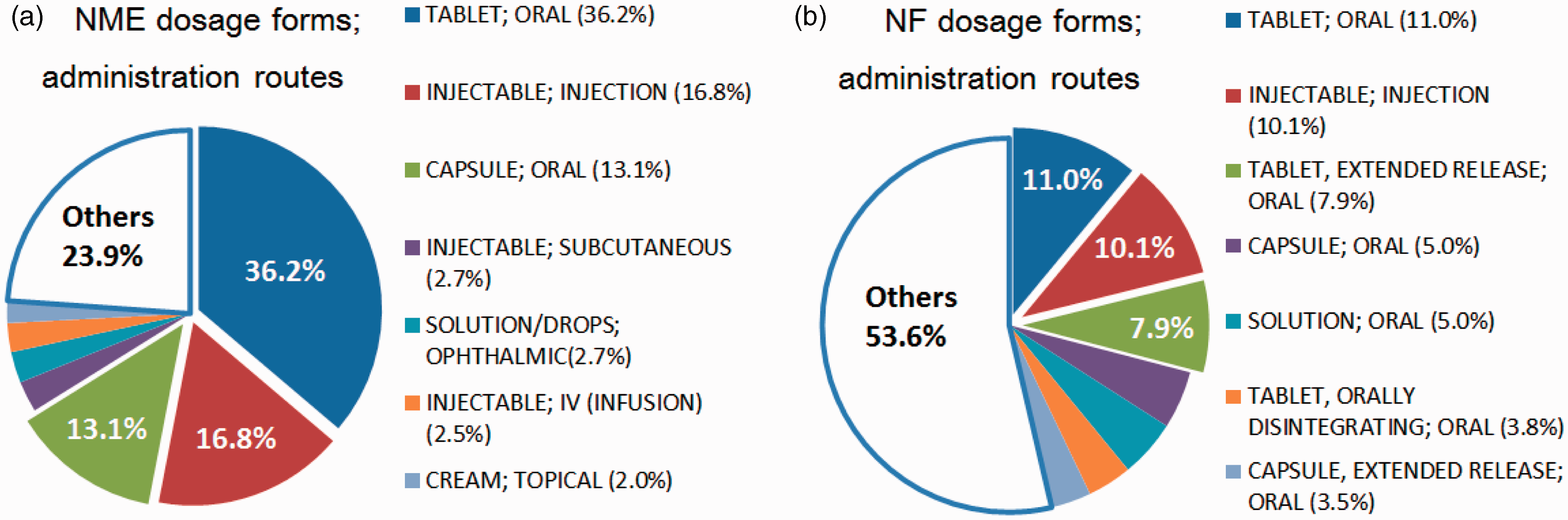
Second, we conduct a survival analysis to measure the LEP for all 180 NF approvals. Figure 4(a) shows the results of the LEP analysis for the “Oral” and “Non-Oral” formulation categories. Chronological changes in survival probability show a significant difference between “Oral” and “Non-Oral” formulations in the median survival at 941 days and 4171 days, respectively.
(a) Kaplan–Meier curves for overall survival by the difference between the curves for “Non-Oral” (n = 56) and “Oral” (n = 124) formulations. A significant difference is observed (log-rank test: p = 0.0003, Wilcoxon test: p = 0.0006). (b) Kaplan–Meier curves for overall survival by the difference between the curves for “Extended/Delayed Release” (n = 38) and “Oral-General” (n = 29) formulations. A significant difference is observed (log-rank test: p = 0.0695, Wilcoxon test: p = 0.0116). (c) Kaplan–Meier curves for overall survival by the difference in oral formulations between the curves for “Patent ≥ 1” (n = 49) and “Patent = 0” (n = 75). A significant difference is observed (log-rank test: p = 0.0171, Wilcoxon test: p = 0.0034). (d) Kaplan–Meier curves for overall survival by the difference in oral formulations between the curves for “Patent = 1” (n = 27), “Patent = 2” (n = 9) and “Patent ≥ 3” (n = 13). No significant differences are observed (log-rank test: p = 0.5319, Wilcoxon test: p = 0.1913).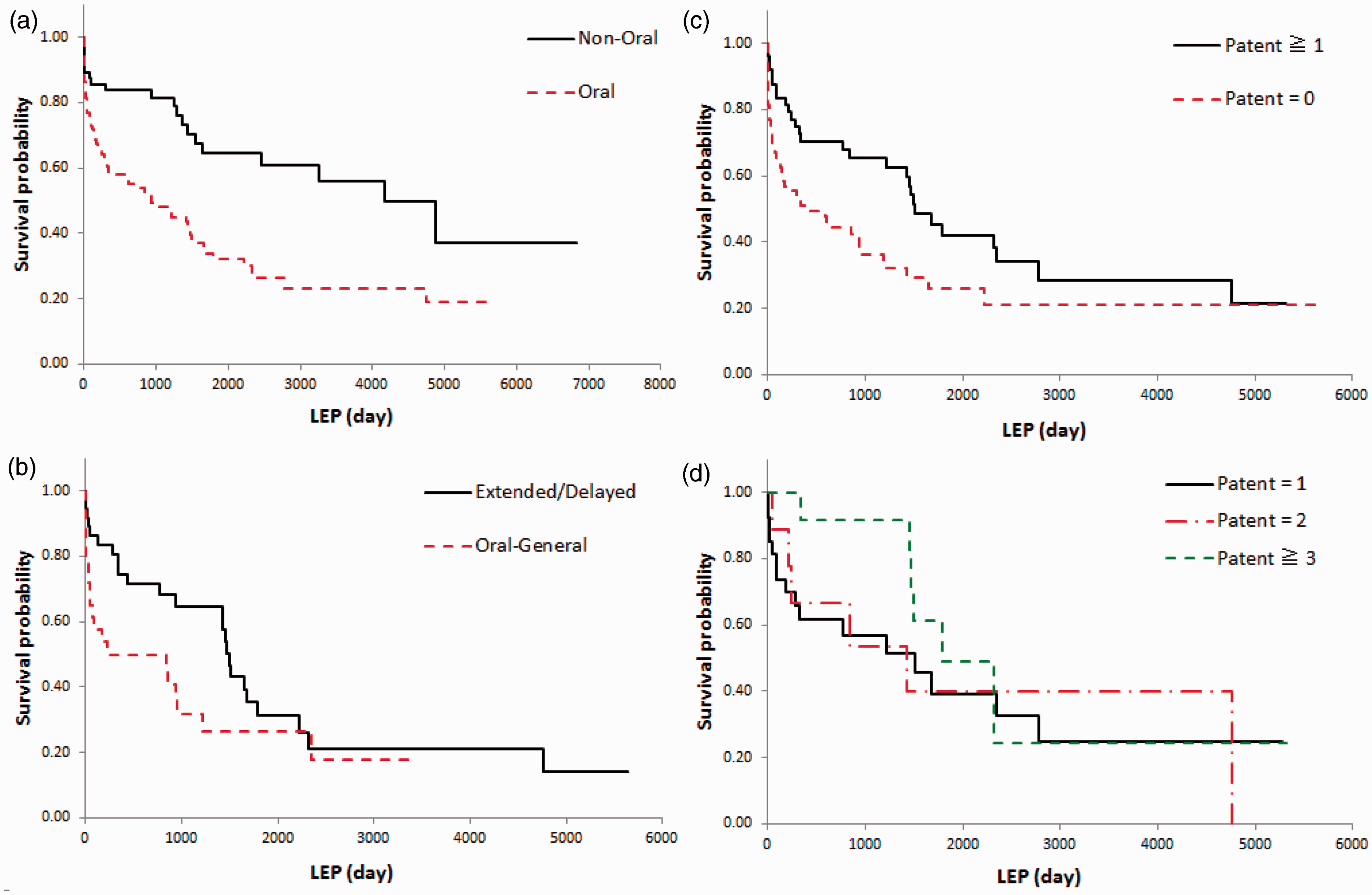
Third, we perform an analysis to investigate the trend of oral dosage forms preventing generic drug entry in the long term and to determine the effects that NF dosage forms have on LEPs. The results show that among the oral formulations, dosage forms such as “Extended/Delayed Release,” which require advanced technology, significantly extend the LEP (Figure 4(b)). Chronological changes in survival probability show a significant difference between “Oral-General” and “Extended/Delayed Release” formulations in the median survival at 229 days and 1498 days, respectively. No significant differences were observed between “Other Oral” and the abovementioned two categories; however, the LEP of “Other Oral” formulations in the median survival was 605 days (not shown in Figure 4).
Fourth, we undertake an analysis to determine the extent to which the acquisition of formulation-related patents affects the LEP within the “Oral” formulation market, where competition between innovator drugs and generic drugs is particularly fierce (Figure 4(c)). Formulation-related patents were identified based on the Orange Book for the years 1995 to 2011 by deleting those patents that protect NME products from the list of patents that protect NF products. Oral formulations (n = 124) were categorized as “Patent ≥1” (n = 49) or “Patent = 0” (n = 75). The former group means that one or more formulation-related patents were listed in Orange Book. The latter group means that no formulation-related patents were listed in that. Kaplan–Meier curves for overall survival by the difference in oral formulations between the curves for “Patent ≥ 1” and “Patent = 0” are illustrated in Figure 4(c). Chronological changes in survival probability show a significant difference between “Patent = 0” and “Patent ≥ 1” formulations in the median survival at 442 days and 1505 days, respectively.
Finally, we attempt a survival analysis to investigate the extent to which the number of formulation-related patents that protect NF products affects the LEP for “Oral” formulations (Figure 4(d)). We subdivided “Patent ≥1” (n = 49) into three groups, “Patent = 1” (n = 27), “Patent = 2” (n = 9), and “Patent ≥3” (n = 13). Each group was given a group name corresponding to the number of formulation-related patents listed in Orange Book. Kaplan–Meier curves for overall survival by the difference in oral formulations between the curves for “Patent = 1”, “Patent = 2”, and “Patent ≥ 3” are illustrated in Figure 4(d). In this case, the results for these numbers of patents (1, 2, and ≥ 3) do not show significant differences with regard to survival probability.
Discussion
This study is the first to attempt a quantitative evaluation of the effects of dosage forms on LEPs by focusing on NF strategies adopted by innovator pharmaceutical companies over the past 20 years.
Top seven categories of NMEs and NFs
The results of the top seven categories of NME and NF formulations in Figure 3 indicate that in an attempt to gain early market share, the NME strategy of innovator pharmaceutical companies is to get pharmaceuticals to the market as quickly as possible, using “Oral-General Formulation” dosage forms typified by tablets and capsules. Tablets are easy to handle and consume and have excellent portability; indeed, they are the most favored formulation. Furthermore, since no special technique is required for manufacturing tablets, the development period is relatively short. However, general formulations such as tablets and capsules hardly serve as technical barriers blocking generic companies. Conversely, it can be also assumed the NF strategy is to use a variety of special formulations that aim to improve the convenience or reduce the adverse effects of using the drug, in an attempt to maximize its value and extend its shelf life.
Survival analysis
Chronological changes in survival probability show a significant difference between “Oral” and “Non-Oral” formulations in the median survival. This may be attributed to the following reason. In contrast to ANDAs for “Oral” generic pharmaceuticals, which can receive approval based solely on bioavailability and bioequivalence (BA/BE) study data, ANDAs for “Non-Oral” generic pharmaceuticals may not be approved unless certain conditions, such as the implementation of separate clinical trials or the use of same additive ingredients as the innovator drug, are met. The differences observed between the survival probabilities of “Oral” and “Non-Oral” formulations were thought to be owing to the effects of such special factors that make it difficult for generic pharmaceutical companies to enter the market.
Even within the “Oral” formulation sector, as described above, “Extended/Delayed Release” formulations exhibit a trend of preventing the market entry of generic drugs in the long term. Dubey and Dubey 8 discussed that while “Oral-General” formulations are more likely to allow generic pharmaceutical companies to make a rapid market entry owing to the low technical hurdles, dosage forms such as “Extended/Delayed Release” formulation, with higher technical hurdles, are the preferred option. However, they also noted that the technical hurdles of “Extended/Delayed Release” formulations have been lowered in recent years because of significant progress in formulation technology and its saturation. This study is the first to focus on NF strategies adopted by innovator pharmaceutical companies over the past 20 years, in order to attempt a quantitative evaluation of the effects of dosage forms on the LEPs, especially by comparing the “Extended/Delayed Release” formulation with other dosage forms. The results of this study demonstrate that the high technical hurdles posed by the “Extended/Delayed Release” formulation have effectively functioned to deter the market entry of generic pharmaceutical companies for the past 20 years (Figure 4(b)). During this time, the acquisition of formulation-related patents for the “Extended/Delayed Release” formulation and the “Oral-General” formulation accounted for about 60% and 20% of the each total approval, respectively, in our data, thus showing a higher rate of patent acquisition for “Extended/Delayed Release” formulation. Additionally, patent acquisitions for “Extended/Delayed Release” formulation were maintained at a high rate of around 60% during the past 20 years for NF approvals, and there is no evidence indicating that technical hurdles for “Extended/Delayed Release” formulations have decreased in recent years. This suggests that formulations with high technical hurdles have made the acquisition of formulation-related patents easier over the past 20 years. Thus, one may consider that technical and patent barriers inherent to “Extended/Delayed Release” formulations could have efficiently blocked the market entry of many generic companies.
In the “Oral” formulation sector, where competition with generic drugs is particularly fierce, the acquisition of formulation-related patents that protect products was observed to extend the life cycle period by approximately 1100 days. This result shows the importance of constructing technical barriers for manufacturing products and of patent barriers to block the market entry of generic companies with NF pharmaceuticals after the RE expires. Few studies have comprehensively evaluated secondary patents, such as formulation-related patents. Kapczynski et al. 17 comprehensively analyzed secondary patents, including formulation-related patents listed in the Orange Book, with a focus on NME pharmaceuticals and indicated that formulation patents add an average of 6.5 years to the nominal exclusivity period of a drug after the expiry of the substance patent. However, their study did not consider the actual timing of the market entry by generic drugs, and thus, lacked a quantitative discussion regarding the actual effects of formulation-related patents on life cycle extension. To date, no experimental studies have focused on the life cycle of NF pharmaceuticals and the timing of generic drug entry without selection bias by excluding censored cases. Therefore, to the best of our knowledge, this study is the first to prove that formulation-related patents, which protect NF pharmaceuticals, contribute to the extension of life cycle periods. Deeper analysis focusing on the number of patents protecting NF products revealed that the number of patents in itself is not so effective at preventing generic drug entry. Therefore, this is the first study to investigate the effects of the number of formulation-related patents on NF pharmaceuticals. The results suggest the importance of the nature of patent barriers, rather than the number of patents, in suppressing the development of circumvention technologies.
However, a difference in survival probability is evident between patent ≥ 3 and patent = 1, 2 in the earlier half of the graph (Figure 4(d)). There is a possibility of change in the Wilcoxon test value should the number of analyzed objects increase. Another limitation of this study is the statistical bias arising owing to the fact that chronological changes in patent numbers were treated as being constant during the observation period.
Summary and conclusion
This study focused on quantifying the LEPs of NF pharmaceuticals by conducting a survival analysis of all samples, including censored cases. The following results were obtained. “Non-oral” formulations greatly extend the LEP, formulation-related patents in the “Oral” formulation sector significantly extend the LEP regardless patent numbers, and “Extended/Delayed Release” formulations, recognized to possess high technical barriers, effectively prevent the entry of generic drugs. Thus, this study proved the importance of non-oral formulation development to delay the market entry of generic drugs more effectively compared to oral formulations. Moreover, it is vital to acquire high quality patents for formulation techniques that pose considerable technical barriers to perfect, so as to protect products in NF strategies. Generic pharmaceutical companies would need to adapt to such approaches of innovator pharmaceutical drug companies and examine future counter strategies.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
None declared.