
Abstract
Osteonecrosis of the femoral head (ONFH) is a progressive, multifactorial bone disease characterized by ischemia-induced osteocyte death, microenvironmental imbalance, and failed tissue regeneration. According to recent advances in pathophysiological understanding, vascular injury, oxidative stress, and inflammatory storms form a pathogenic cascade leading to osteogenic dysfunction, adipogenic lineage drift of mesenchymal stem cells (MSCs), and epigenetic alterations that exacerbate bone degeneration. Despite improvements in early detection, traditional interventions—including bisphosphonates, hyperbaric oxygen therapy, and surgical decompression—have limited efficacy, particularly in the mid-to-late stages. This review systematically synthesizes emerging regenerative approaches across three domains: (1) Cellular and molecular therapies: Autologous MSC transplantation, exosomes, and apoptotic extracellular vesicles restore osteogenesis, modulate immunity, and promote angiogenesis, while gene-editing technologies such as CRISPR/Cas9 enhance MSC functionality. (2) Nanobiomaterial synergy: Enzyme-mimetic nanozymes and multifunctional polymeric scaffolds improve lesion targeting, reactive oxygen species clearance, and microenvironmental regulation. (3) Advanced bioengineering: Organoid models and 3D-bioprinted living joint prostheses enable the integration of vascularization, mechanical support, and precise drug delivery, representing transformative strategies in personalized repair. Together, these innovations highlight a future paradigm shift from passive support to active, mechanism-targeted regeneration, offering new hope for structural and functional reconstruction in ONFH. Multidisciplinary integration—bridging materials science, stem cell biology, and digital medicine—will be essential for successfully developing functional cures.
Impact Statement
This review articulates a paradigm shift in osteonecrosis of the femoral head management, from palliative care to proactive, mechanism-driven regeneration. By integrating breakthroughs in cell therapy, nanobiomaterials, and bioengineering, it charts a path for true structural and functional joint restoration. These integrated strategies—precisely targeting the pathogenic triad of ischemia, oxidative stress, and inflammation—hold the transformative potential to halt disease progression and achieve biological joint reconstruction.
Introduction
Osteonecrosis of the femoral head (ONFH), also known as avascular necrosis, is a debilitating condition caused by impaired blood supply to the femoral head, leading to osteocyte death and failure of bone repair. It typically progresses from subclinical ischemia to subchondral collapse and secondary osteoarthritis, often requiring total hip arthroplasty (THA). Globally, ONFH affects about 30 million people, with over 8.1 million nontraumatic cases in China and 20,000–30,000 new annual cases in the United States.1–5 Without early intervention, 70–80% of patients progress to end-stage joint degeneration, creating a significant clinical and socioeconomic burden. 6 The pathogenesis centers on vascular injury due to traumatic causes (e.g., femoral neck fractures) or nontraumatic factors such as glucocorticoid use (∼51%) and alcohol abuse (∼31%).5,7,8 These lead to bone homeostasis disruption via lipid dysregulation, suppressed osteogenesis, microvascular dysfunction, and proapoptotic effects.9–11 Early ischemia triggers apoptosis and necrosis, and once femoral head collapse exceeds 2 mm, joint preservation becomes unfeasible. 6
Treatment includes conservative methods (bisphosphonates, statins, hyperbaric oxygen) and joint-preserving surgeries (core decompression [CD], bone grafting), though their efficacy is limited in advanced stages. THA remains the definitive solution for late-stage ONFH. Recent advances in single-cell RNA sequencing, spatial transcriptomics, and regenerative approaches—such as stem cell therapy, gene editing, and 3D bioprinting—offer new insights into the disease microenvironment and potential for functional regeneration, shifting the paradigm from structural repair to biologically guided reconstruction. This review synthesizes molecular mechanisms and evolving treatments in ONFH, highlighting translational strategies for future precision medicine and joint preservation.
Pathogenesis of ONFH
Vascular injury and hypoxic microenvironment
Vascular injury is a central pathogenic mechanism in ONFH and is driven by a multifactorial interplay of structural vascular disruption, hemodynamic abnormalities, and impaired neovascularization. Reduced perfusion of the femoral head can result from several interrelated etiological mechanisms: (1) Direct vascular trauma: Fractures of the femoral neck, hip dislocations, or iatrogenic surgical injury can compromise branches of the medial and lateral femoral circumflex arteries, acutely disrupting blood flow to the femoral head. 12 (2) Intramedullary hypertension: Conditions such as sickle cell anemia increase intraosseous pressure, whereas glucocorticoid exposure and chronic alcohol consumption stimulate adipogenic differentiation and lipid accumulation in bone marrow mesenchymal stem cells (MSCs) via upregulation of peroxisome proliferator-activated receptor gamma (PPARγ). This leads to adipocyte hypertrophy, marrow edema, and increased intramedullary pressure, resulting in the compression of intraosseous microvessels and further vascular compromise.10,13–16 (3) Intravascular occlusion: Hypercoagulable states associated with inherited or acquired thrombophilia (e.g., factor V Leiden mutation; prothrombin gene G20210A mutation; and deficiencies in antithrombin III, protein C or S, or the MTHFR C677T gene polymorphism) predispose patients to thrombotic occlusion of femoral head vessels. 17 In addition, fat embolism secondary to dyslipidemia or gas emboli can further impair blood flow. 18 (4) Endothelial apoptosis: Glucocorticoid (GCs) promote endothelial cell death through the dysregulation of pro and antiapoptotic signaling and induction of endoplasmic reticulum (ER) stress, leading to microvascular degradation and subsequent tissue ischemia. 19 (5) Impaired angiogenesis: GCs suppress the expression of vascular endothelial growth factor (VEGF) and reduce both the number and functional capacity of endothelial progenitor cells, inhibiting migration and vascular repair while promoting cellular senescence.20–22
Beyond these primary mechanisms, additional vascular pathologies, such as vasoconstriction and atherosclerosis, contribute to impaired femoral head circulation.23,24 These insults act synergistically rather than independently, culminating in irreversible osteocyte death and subchondral collapse. Given the complexity of these interacting pathways, future therapeutic strategies must move beyond single-target interventions.
Oxidative stress and inflammation in ONFH
Oxidative stress and chronic inflammation represent interlinked and pivotal mechanisms in the pathogenesis of ONFH. The major oxidative stress-related signaling pathways involved in ONFH are illustrated in Figure 1. Exogenous insults—most notably glucocorticoid overuse, excessive alcohol intake, and aberrant mechanical loading—promote the overproduction of reactive oxygen species (ROS), thereby disrupting redox homeostasis and establishing a persistent oxidative microenvironment within the femoral head.25,26
NADPH oxidases (NOXs), particularly NOX1, NOX2, and NOX4, are major enzymatic sources of ROS in skeletal tissue.27,28 In vitro, glucocorticoids upregulate NOXs in both bone marrow mesenchymal stem cells (BMSCs) and MC3T3-E1 osteoblast-like cells, leading to increased ROS production, impaired osteogenic differentiation, and cell apoptosis.29,30 Central to the cellular antioxidant defense system is the Keap1/Nrf2 signaling axis, in which nuclear factor erythroid 2-related factor 2 (Nrf2) regulates the transcription of detoxifying and ROS-scavenging enzymes. GCs disrupt this axis through multiple pathways.29,31 In addition, GCs downregulate the expression of other antioxidant enzymes in necrotic bone tissue, further promoting ROS formation and enhancing oxidative stress. 32 ROS can stimulate the mitogen-activated protein kinase (MAPK) pathway, increasing the phosphorylation of ASK1 and p38 in cells, thereby inducing cell apoptosis and accelerating cartilage matrix degradation.30,33
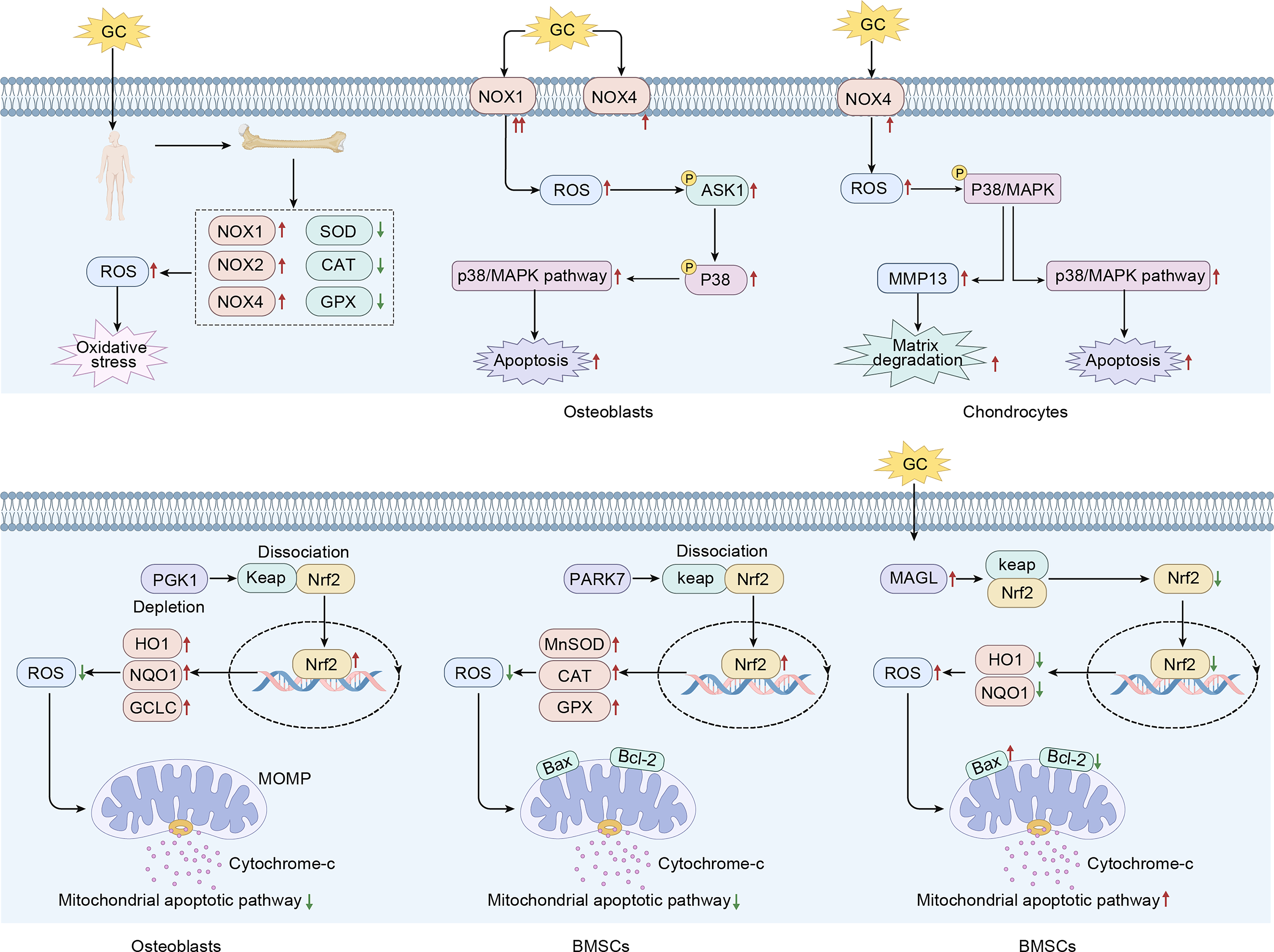
The role of oxidative stress in the pathogenesis of ONFH. Glucocorticoids can act on the NOX protein family, resulting in increased generation of reactive oxygen species (ROS). Through the P38/MAPK pathway, this leads to enhanced apoptosis and promotes the degradation of the cartilage matrix. In femoral head necrosis, the Keap/Nrf2 signaling pathway plays a crucial regulatory role in oxidative stress, and its upstream molecules, including PGK1, PARK7, and Monoacylglycerol Lipase (MAGL), are further involved in the regulation of the Keap/Nrf2 signaling pathway. MAPK, mitogen-activated protein kinase; NOX, NADPH oxidase; ONFH, osteonecrosis of the femoral head.
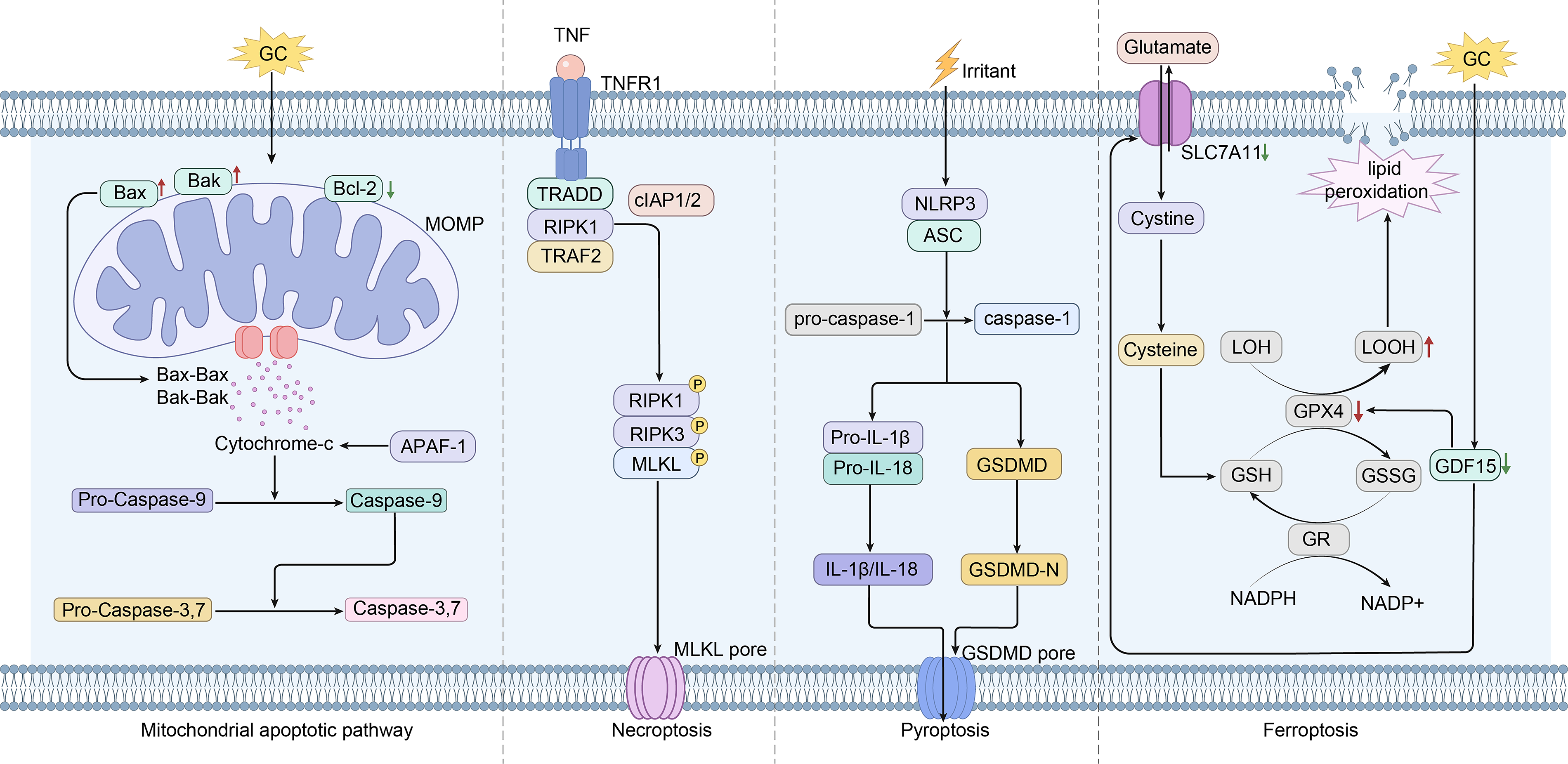
Flowchart of cell death modes in the progression of femoral head osteonecrosis. Flowchart illustrating the distinct cell death modalities, including apoptosis, necroptosis, pyroptosis, and ferroptosis, during the progression of femoral head necrosis. ONFH involves multiple regulated cell death pathways. Apoptosis is triggered via the mitochondrial pathway, characterized by Bax upregulation, Bcl-2 downregulation, and caspase-3 activation. Necroptosis occurs through RIPK1/RIPK3/MLKL phosphorylation, promoting inflammatory death in bone microvascular endothelial cells. Oxidative damage in osteoblasts results from GPX4 and SLC2A1 downregulation alongside TFR1 and NCF2 upregulation. Ferroptosis in mesenchymal stem cells is mediated by the GDF15/SLC7A11-GSH-GPX4 axis, depleting intracellular antioxidants. In addition, pyroptosis is indicated by upregulated NLRP3, Gasdermin D (GSDMD), and caspase-1, activating inflammasome signaling. ONFH, osteonecrosis of the femoral head.
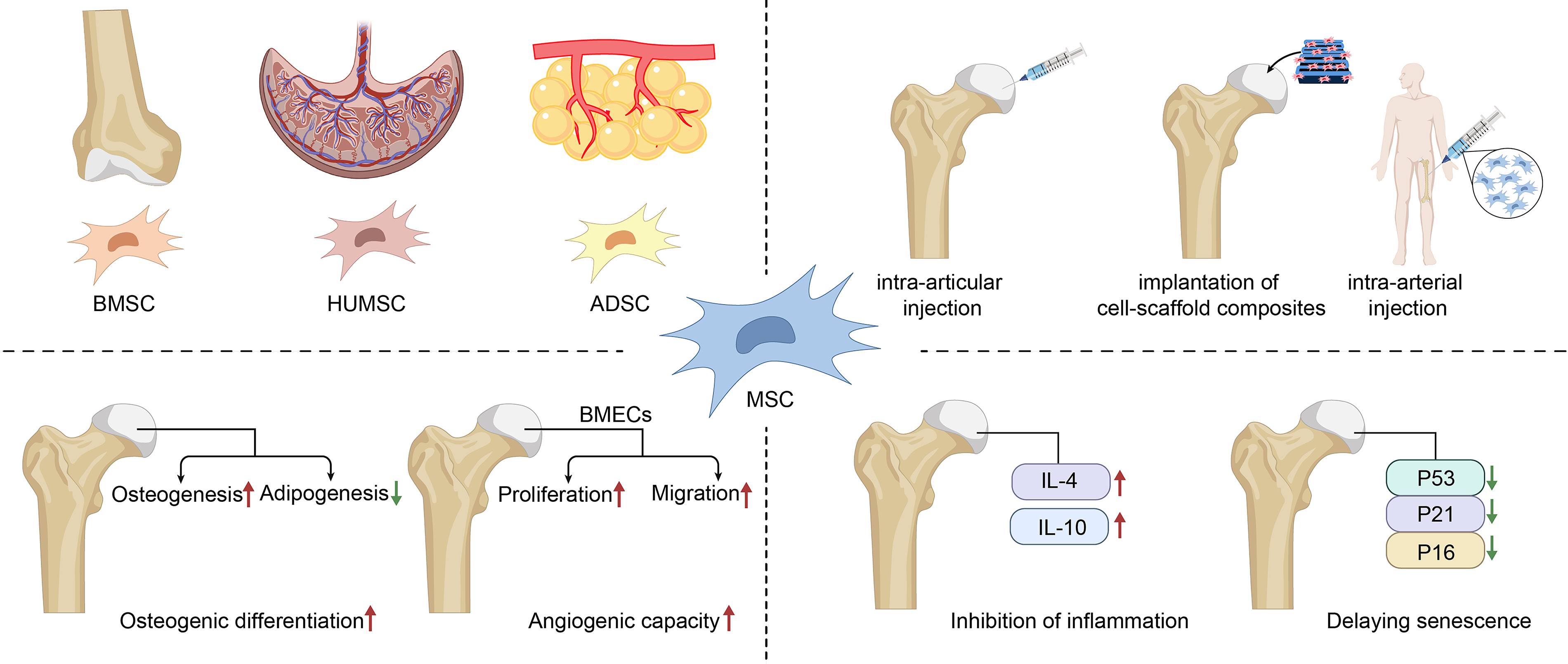
Mesenchymal stem cell-based therapies for osteonecrosis of the femoral head. Extensive research on mesenchymal stem cells derived from various tissues in osteonecrosis of the femoral head has been carried out. In the clinical management of ONFH, MSCs can exert therapeutic efficacy through multiple administration approaches, including intra-articular injection, combined scaffold implantation, and arterial injection. Mechanistically, MSCs can attenuate the progression of ONFH by facilitating osteogenic differentiation, promoting angiogenesis, suppressing inflammatory responses, and delaying cellular senescence. MSCs, mesenchymal stem cells; ONFH, osteonecrosis of the femoral head.
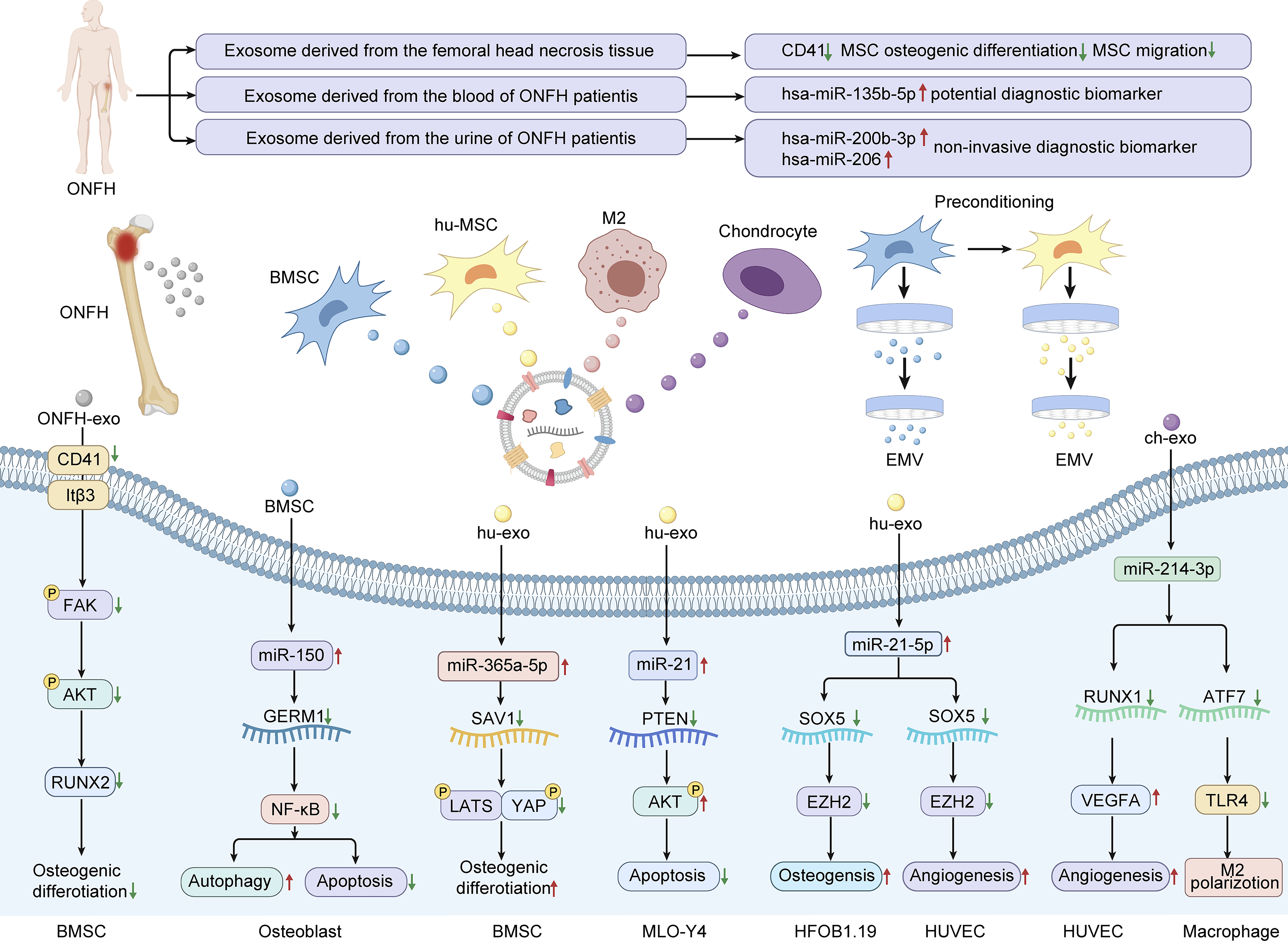
The role of exosomes in the diagnosis and disease progression of patients with ONFH. Pathologically derived exosomes (ONFH-Exos) from necrotic bone tissue exacerbate glucocorticoid-induced osteonecrosis by downregulating CD41 expression, thereby impairing mesenchymal stem cell migration and osteogenic differentiation. Diagnostically, serum exosome levels in steroid-induced patients with ONFH are significantly lower than in healthy controls. Elevated hsa-miR-135b-5p in serum exosomes serves as a potential early biomarker, while increased urinary exosomal hsa-miR-200b-3p and hsa-miR-206 suggest a noninvasive approach for early ONFH detection. The preparation of exosomes derived from various cell types and exosome mimetics, as well as the exploration of the mechanisms underlying the therapeutic effects of exosomes in ONFH. ONFH, osteonecrosis of the femoral head.
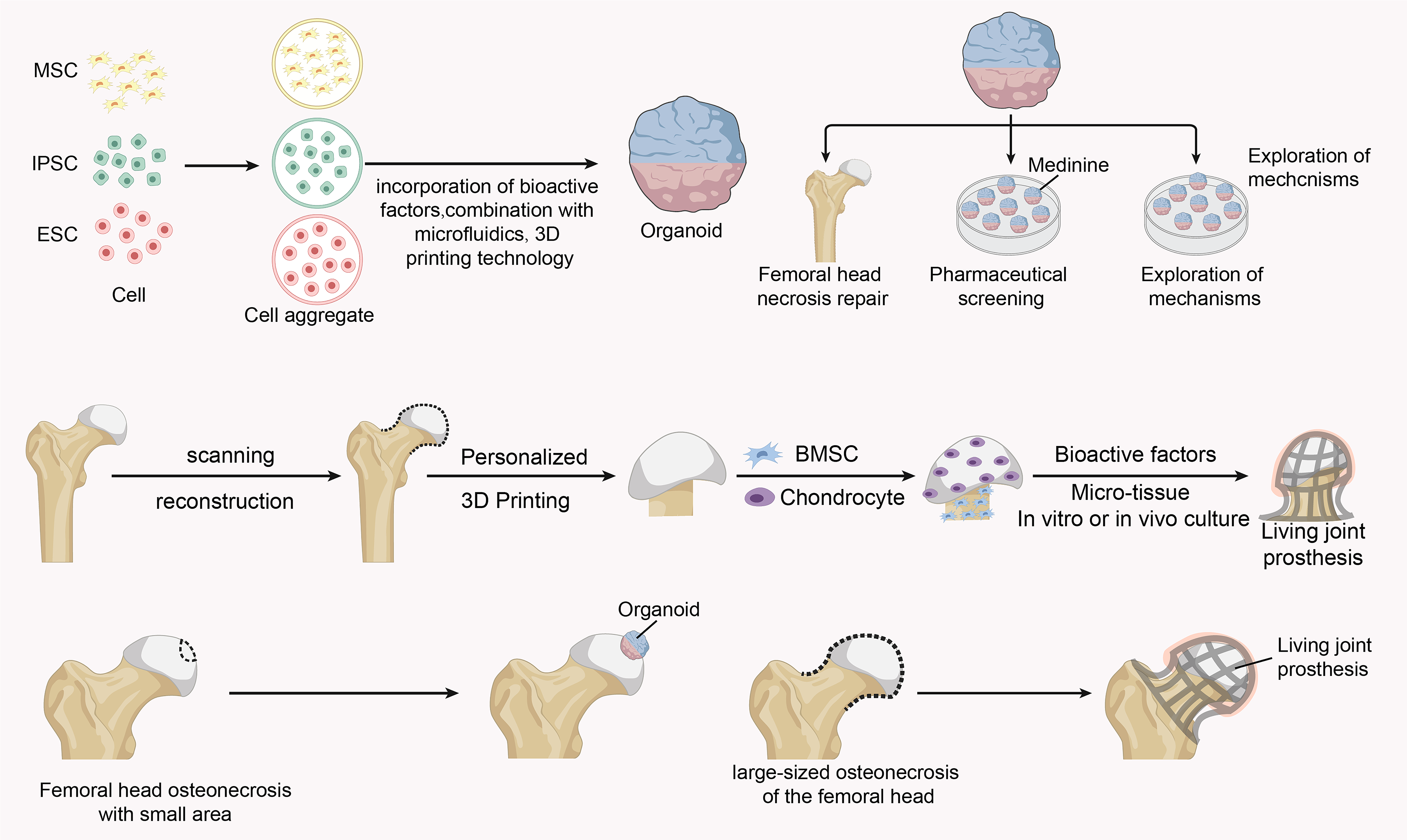
The potential role of organoid-based modules in osteonecrosis of the femoral head. Compositional profiles and repair potential of femoral head organoids and bioactive joint prostheses, as well as the potential of organoids in drug screening and pathogenesis exploration in osteonecrosis of the femoral head.
Summary of Different Modes of Cell Death in the Femoral Head
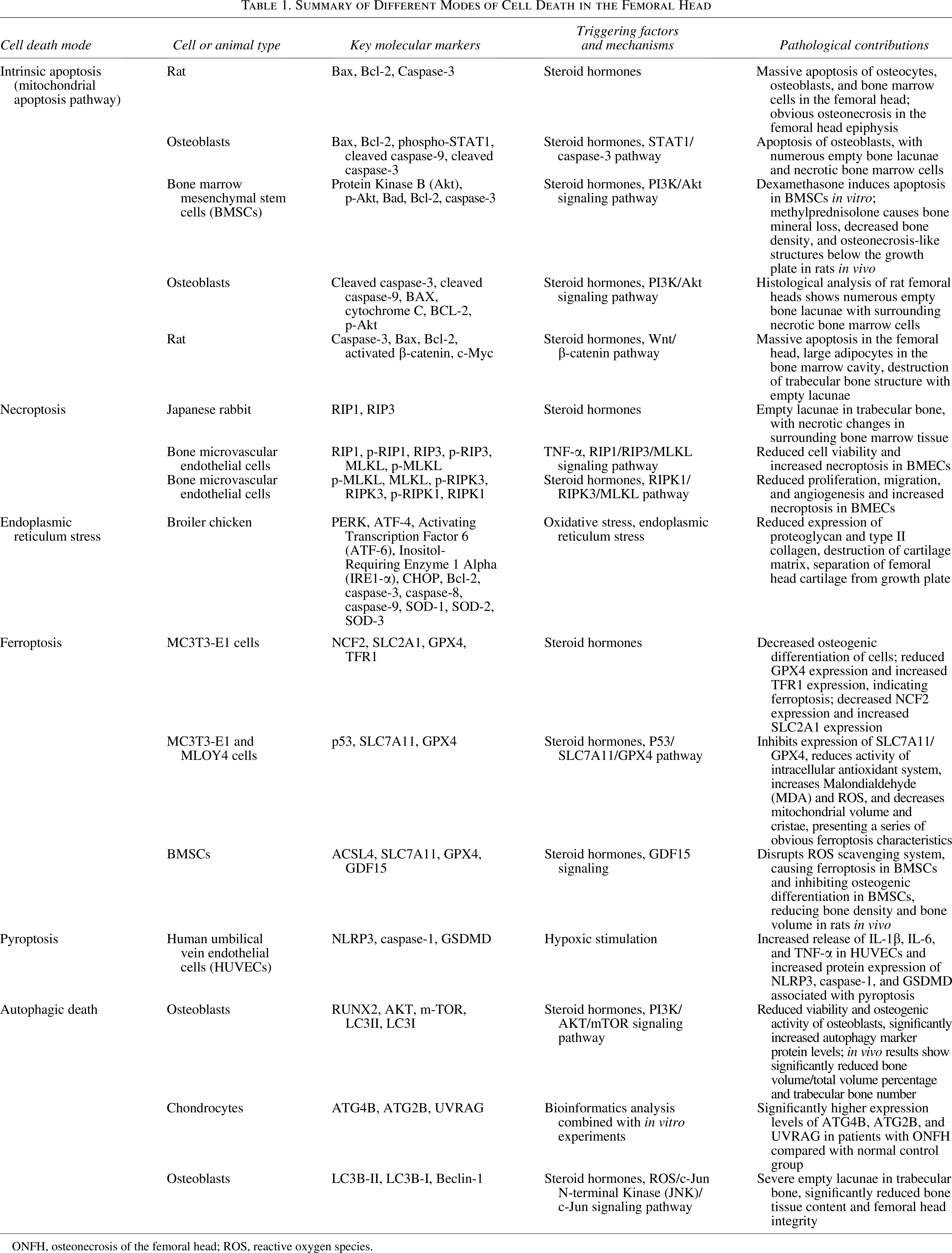
ONFH, osteonecrosis of the femoral head; ROS, reactive oxygen species.
Classification of Epigenetic Alterations in Osteonecrosis of the Femoral Head
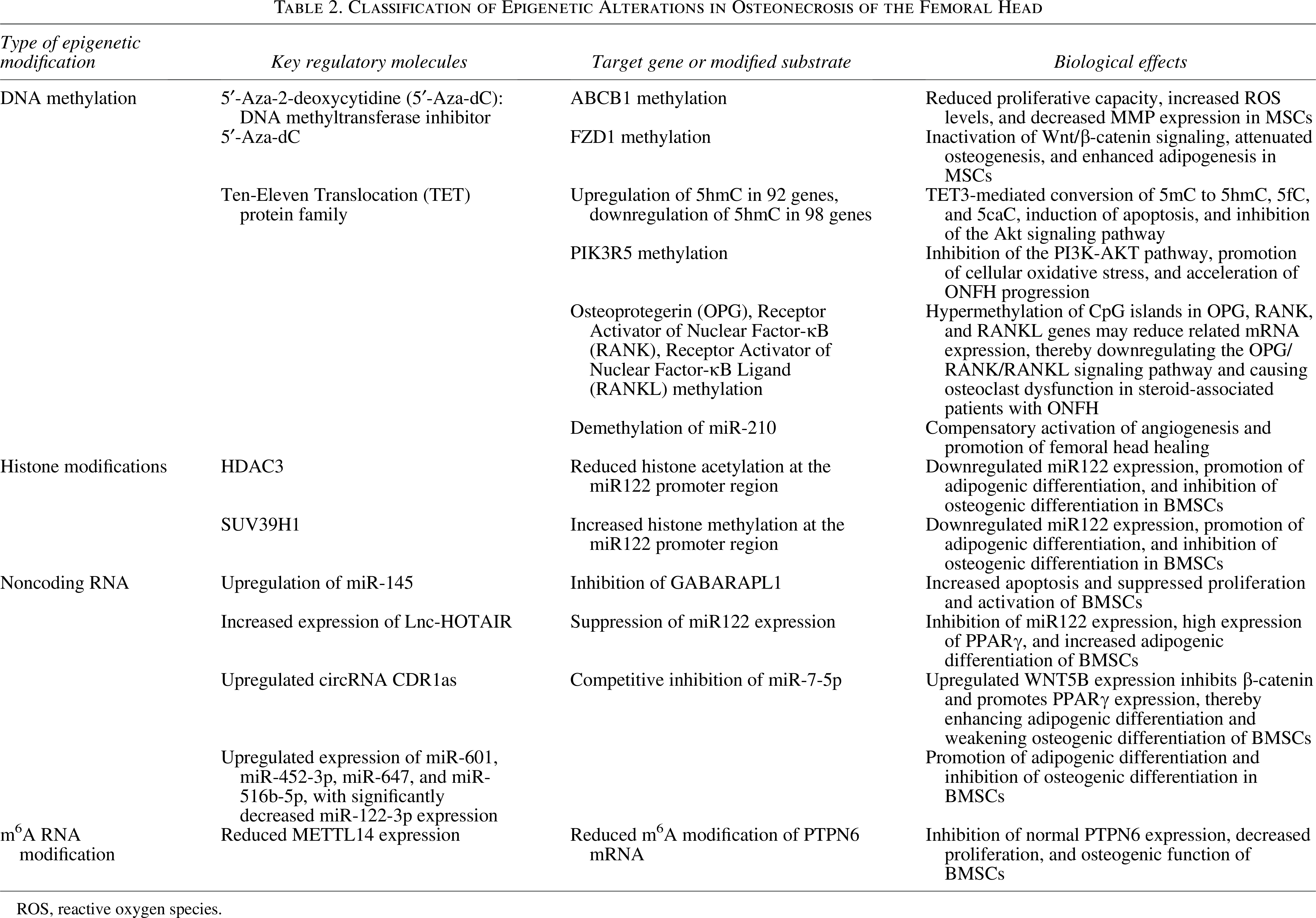
ROS, reactive oxygen species.
Application of Conventional Therapeutic Approaches in Osteonecrosis of the Femoral Head

VEGF, vascular endothelial growth factor.
Summary of the Therapeutic Mechanisms of Exosomes in Osteonecrosis of the Femoral Head (ONFH) and Disease Progression
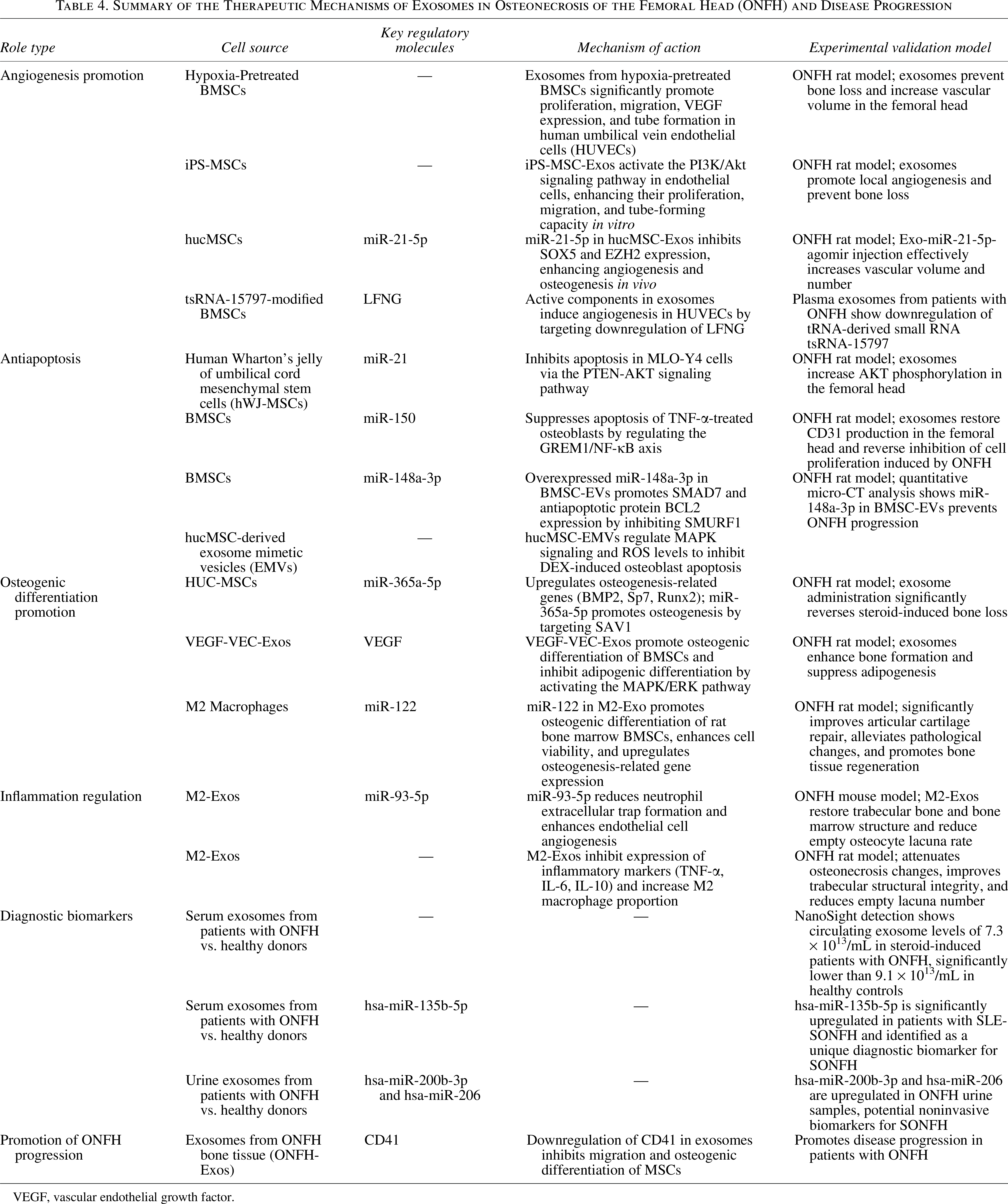
VEGF, vascular endothelial growth factor.
Roles of Polymers in the Treatment of Osteonecrosis of the Femoral Head
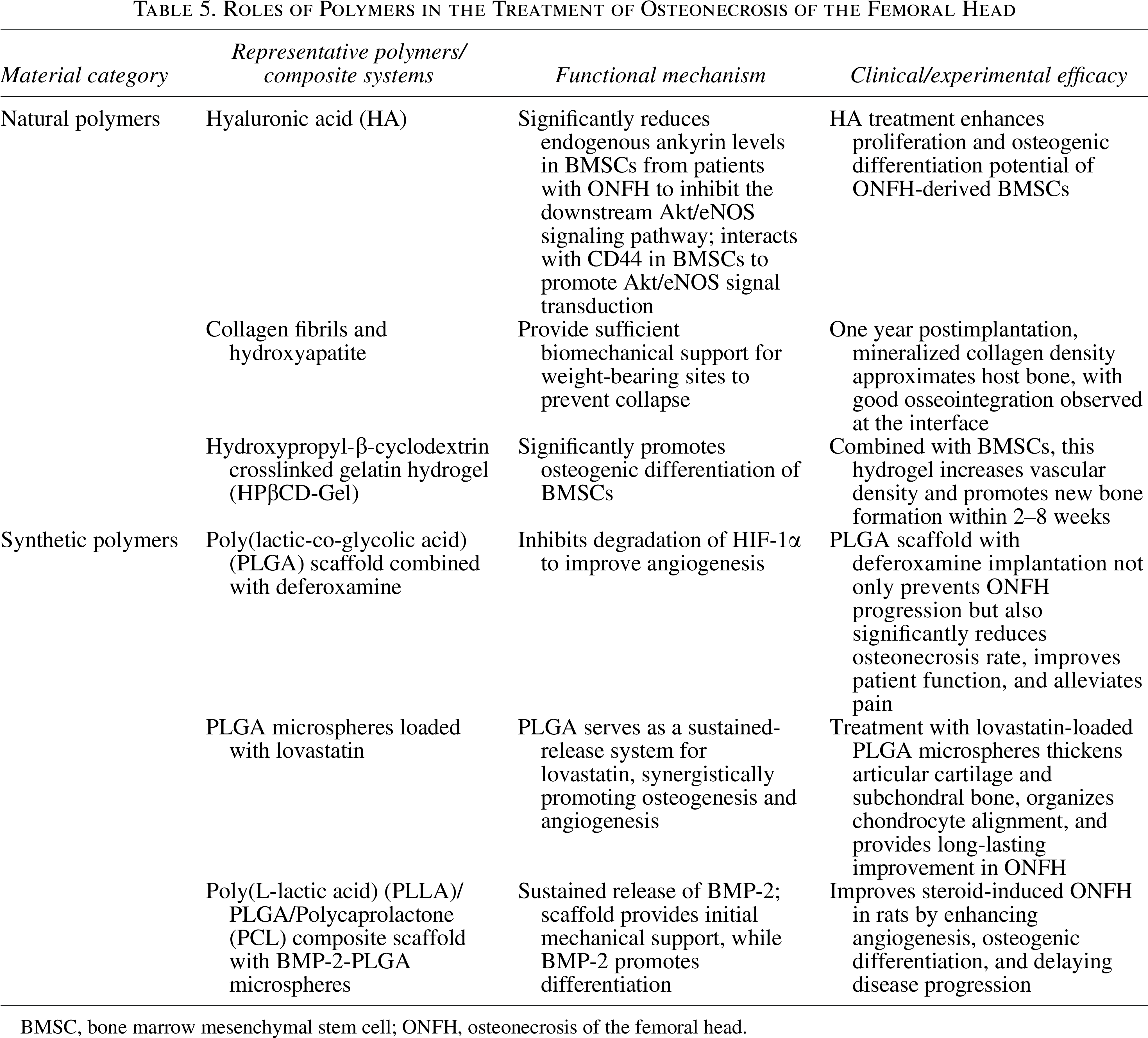
BMSC, bone marrow mesenchymal stem cell; ONFH, osteonecrosis of the femoral head.
Persistent ROS accumulation impairs osteogenesis by inhibiting the expression of transcription factors such as Runx2, Osterix (Osx), and Dlx5 while simultaneously promoting osteoclastogenesis via upregulation of the expression of tartrate-resistant acid phosphatase, c-Fos, and NFATc1. 34 This dysregulation establishes a pathological imbalance between bone formation and resorption. Moreover, oxidative stress and inflammation are mechanistically linked: ROS can activate tumor necrosis factor (TNF)-mediated nuclear factor kappa B (NF-κB) signaling, leading to elevated expression of proinflammatory mediators, including TNF, NOX2, interleukin-6 (IL-6), and IL-8. 35
As a core regulator of the osteoimmune microenvironment in ONFH, the imbalance in M1/M2 phenotypic polarization of macrophages is a key mechanism driving disease progression—initial insults such as ischemia and oxidative stress induce macrophages to polarize toward the M1 phenotype, which aggravates inflammatory responses, induces apoptosis of vascular endothelial cells, and promotes osteoclast activation by secreting proinflammatory factors including TNF-α and IL-6. In contrast, the reduced proportion of M2 macrophages leads to the loss of their anti-inflammatory and reparative functions (e.g., secretion of IL-10, VEGF, and BMP-2), further exacerbating bone metabolic disorders and vascular damage. Targeted regulation of macrophage phenotypes has emerged as a potential therapeutic strategy. 36 In addition, in patients with ONFH, elevated expression of IL-21 and IL-9 enhances JAK-STAT signaling activation, which further induces the upregulation of inflammation-related cytokines (e.g., TNF-α, IL-6, and COX-2) and catabolic biomarkers (including ADAMTS-4 and MMP-13), ultimately exacerbating ONFH progression.37,38
These insights highlight several potential intervention targets. Inhibiting NOX activity, restoring Nrf2-driven antioxidant defenses, or blocking key inflammatory cascades may offer promising strategies for disrupting the pathological feedback loop and mitigating ONFH progression.
Multiple cell death pathways in ONFH pathogenesis
The pathophysiology of ONFH involves a complex cascade of cellular injury, wherein exogenous insults such as glucocorticoids, alcohol, and mechanical stress induce ischemia, oxidative stress, and inflammation within the femoral head microenvironment. The regulated cell death pathways implicated in ONFH are summarized in Figure 2. These upstream disturbances trigger the aberrant activation of multiple cell death pathways—including apoptosis, necroptosis, ferroptosis, and pyroptosis—which serve as core mediators of bone homeostasis disruption and structural collapse.
(1) Apoptosis, a well-characterized form of programmed cell death, is predominantly mediated through the intrinsic (mitochondrial) pathway. This pathway is initiated by alterations in mitochondrial membrane permeability, resulting in cytochrome c release and caspase activation. In GC-induced ONFH, in vivo and in vitro studies have demonstrated the upregulation of proapoptotic proteins (e.g., Bax), the suppression of antiapoptotic regulators (e.g., Bcl-2), and the subsequent activation of caspase-3.39,40 GCs also induce osteoblast apoptosis via the STAT1/caspase-3 axis, while in BMSCs, mitochondrial pathway activation leads to similar proapoptotic effects.41,42 Several intracellular signaling cascades modulate apoptosis susceptibility: activation of the PI3K/AKT pathway inhibits apoptosis by downregulating Bcl-2-Associated X Protein (Bax) and caspase activation; conversely, inhibition of the Wnt/β-catenin pathway exacerbates apoptosis through upregulation of Bax expression and suppression of Bcl-2 expression.42,43 In addition, the MAPK pathway is involved, as GCs increase JNK and p38 phosphorylation, accelerating apoptosis within the femoral head marrow. 2 In idiopathic ONFH, ER stress contributes to apoptosis by upregulating the expression of C/EBP Homologous Protein and its downstream interactions with Bcl-2 family proteins. 44 (2) Nonapoptotic regulated cell death pathways have recently been confirmed as critical factors in ONFH progression. 45 Under GC and TNF-α stimulation, necroptosis occurs in bone microvascular endothelial cells, further compromising the vascular supply to the femoral head.46,47 (3) Ferroptosis, characterized by iron-dependent lipid peroxidation and mitochondrial dysfunction, is increasingly recognized in steroid-induced ONFH. 48 GC exposure enhances oxidative damage in osteoblasts and triggers ferroptosis in BMSCs.49,50 (4) Pyroptosis, an inflammatory cell death form mediated by Gasdermin family proteins, is also implicated. Activation of the NLRP3 inflammasome and caspase-1 in human umbilical vein endothelial cells (HUVECs) promotes interleukin-1β (IL-1β) secretion, with fibroblast growth factor 23 (FGF23) identified as an upstream regulator. 51 In addition, autophagic cell death has recently gained attention in the context of ONFH.52–54 (5) Moderate autophagy is essential for normal cellular metabolism, whereas excessive autophagy can lead to abnormal cell death in the femoral head. 53 Steroid hormones induce excessive autophagy in osteoblasts via the PI3K/AKT/mTOR and ROS/JNK/c-Jun signaling pathways, upregulating autophagy markers (LC3II/I, Beclin-1) and downregulating osteogenic factors such as RUNX2, thereby impairing osteoblast function. This process results in bone loss, trabecular bone destruction, and compromised structural integrity of the femoral head. Meanwhile, significantly elevated expression of ATG4B, ATG2B, and UVRAG is observed in chondrocytes from patients with ONFH, suggesting that dysregulated autophagy in both osteoblasts and chondrocytes contributes jointly to the pathogenesis of steroid-induced ONFH.52–54
The major cell death modalities involved in ONFH are summarized in Table 1. Collectively, these diverse cell death programs interact dynamically to accelerate ONFH progression. Elucidating the spatiotemporal dynamics and cross-talk among these death networks is critical for identifying actionable molecular targets and guiding the development of precision therapeutic strategies aimed at halting disease progression and preserving femoral head viability.
MSC dysfunction and epigenetic regulation in ONFH
Mesenchymal stem cells are pivotal regulators of bone homeostasis, and their dysfunction significantly contributes to the pathogenesis of ONFH. Studies demonstrate that reduced MSC quantity or impaired osteogenic capacity disrupts the balance of bone formation, resorption, and remodeling. This compromised regenerative potential limits effective compensation for ischemic necrosis, accelerating disease progression. Aberrant intracellular signaling pathways further dysregulate MSC differentiation. For instance, upregulation of COUP-TFII promotes adipogenesis while suppressing osteogenesis, aggravating ONFH. 55 Similarly, activation of the Notch-RBPJ pathway in avian models reduces osteogenic and chondrogenic differentiation, favoring adipocyte commitment. 56
Emerging evidence underscores the central role of epigenetic dysregulation in MSC dysfunction during ONFH. 57 Risk factors like glucocorticoid exposure or alcohol intake induce adipogenic–osteogenic imbalance through alterations in DNA methylation, histone modifications, and noncoding RNA expression.58,59 These changes repress osteogenic gene programs while promoting adipogenesis, thereby weakening bone repair and amplifying pathogenesis through inflammation and lipotoxicity.
DNA methylation is a well-characterized epigenetic mechanism in ONFH. Genome-wide methylation profiling revealed significantly higher methylation levels in ONFH-BMSCs, with 1833 differentially methylated CpG sites identified. 60 Promoter hypermethylation of PIK3R5 downregulates the PI3K–AKT pathway, impairing oxidative stress responses. 61 Hypermethylation of OPG, RANK, and RANKL disrupts the OPG/RANK/RANKL axis, contributing to osteoclast dysfunction in steroid-induced ONFH. 62 CpG island hypermethylation of FZD1 suppresses Wnt/β-catenin signaling, shifting MSC differentiation toward adipogenesis. 63 Ten-eleven translocation proteins also promote osteocyte apoptosis via epigenetic remodeling, 64 while hypermethylation of ABCB1 may underlie interindividual variability in glucocorticoid susceptibility. 65
Histone modifications similarly contribute to ONFH pathogenesis. In ethanol-induced models, lncRNA HOTAIR overexpression suppresses miR-122 and upregulates PPARγ, promoting BMSC adipogenesis. Mechanistically, PPARγ recruits HDAC3 and SUV39H1 to the miR-122 promoter, reducing histone acetylation and increasing methylation to maintain suppression. 59
Noncoding RNAs critically modulate MSC fate.66,67 Upregulated miR-145 inhibits GABARAPL1, whereas its inhibition enhances BMSC proliferation and reduces apoptosis. 68 Dysregulation of other miRNAs—including upregulated miR-601, miR-452-3p, miR-647, and miR-516b-5p and downregulated miR-122-3p—promotes adipogenesis over osteogenesis. 69 Additional expression alterations involve upregulated miR-4440 and downregulated miR-6787-5p, miR-500a-3p, miR-130a-3p, miR-3663-3p, and miR-222-3p, with GO and KEGG analyses confirming their pivotal roles. 70
Representative epigenetic alterations associated with ONFH are summarized in Table 2. Notably, some epigenetic alterations may mediate compensatory responses. Clinical analyses show miR-210 upregulation in necrotic regions, and subsequent studies confirm that miR-210 demethylation enhances its expression, promotes angiogenesis, and facilitates femoral head repair. 71 These findings indicate that epigenetic reprogramming not only drives ONFH pathogenesis but also offers therapeutic targets for restoring osteogenic capacity and vascular regeneration.
Current Therapeutic Strategies
Hyperbaric oxygen and shock wave therapies in ONFH
Hyperbaric oxygen therapy (HBOT) improves tissue oxygenation, reduces intraosseous pressure, and promotes angiogenesis in early ONFH (Steinberg I–II), with response rates up to 64%.72,73 HBOT upregulates osteoprotegerin, modulates the OPG/RANKL ratio, and suppresses proinflammatory cytokines (IL-6, TNF-α) and NF-κB signaling.74–76 Extracorporeal shock wave therapy (ESWT) demonstrates efficacy in ARCO I–II ONFH, improving Harris Hip Scores (77.4–86.9) and reducing pain. 77 ESWT promotes angiogenesis via VEGF, vWF, and PECAM-1 upregulation, and enhances osteogenesis through Wnt pathway activation and BMP-2/VEGF expression.78–80
Pharmacologic therapies
Bisphosphonates inhibit osteoclast activity and prevent osteocyte apoptosis. While some studies show reduced femoral head collapse (2/29 vs. 19/25 in controls), others report no significant benefit,81–83 highlighting the need for stage-specific application. Statins exert pleiotropic effects by activating Wnt signaling through the BMSC WNT5A/LRP5 axis, promoting osteogenesis (Runx2, Alpl) and suppressing adipogenesis (Pparg, Cebpb).84–86 Pravastatin additionally reduces oxidative stress and promotes angiogenesis. 85 Vasodilators and anticoagulants (alprostadil, iloprost, LMWH) improve microcirculation and prevent thrombosis. LMWH delays disease progression and reduces THA requirements, particularly in patients with thrombophilia.87–90
Joint-preserving surgical strategies
CD reduces intraosseous pressure and creates revascularization channels, achieving 79% success in Ficat stage I.91,92 Multiple small-diameter drilling preserves trabecular structure better than single-core techniques. 93 Enhanced by navigation systems and augmented reality, CD combined with BMSC transplantation or tantalum rods improves outcomes.94–98 Bone grafting provides mechanical support and osteogenic stimulation. 99 Vascularized bone grafting (VBG) shows durable results in Ficat II–III patients.100,101 Innovative approaches include β-TCP bioceramics (88.25% hip survival) and magnesium screw fixation (95.7% success).102,103 Gene-modified stem cells (e.g., CGRP-BMSCs) further enhance bone regeneration. Conventional therapeutic approaches for ONFH are summarized in Table 3. For end-stage osteoarthritis (OA), joint replacement constitutes the conventional surgical treatment strategy. 104
Emerging Therapeutics
Mesenchymal stem cells in ONFH: Multifaceted regenerative and immunomodulatory roles
MSCs originate from mesodermal mesenchymal tissue and have been extensively applied in the treatment of musculoskeletal disorders, including bone defects, cartilage degeneration, osteoarthritis, and, notably, ONFH.105,106
Therapeutic mechanisms of MSC-based interventions in ONFH are illustrated in Figure 3. Clinical studies have demonstrated that local autologous bone marrow-derived MSC (BMSC) injection significantly improves clinical outcomes in patients with ONFH. For instance, patients receiving intrafemoral BMSC therapy exhibited marked increases in both pain relief and hip function scores, with mean Harris Hip Scores increasing from 52.00 ± 18.02 to 67.00 ± 27.55. Meanwhile, Magnetic Resonance Imaging (MRI)-based assessments of femoral head structure also showed improvements at the 6-month follow-up. 107
Preclinical models further confirm the therapeutic efficacy of MSCs: MSC therapy has also been associated with increased serum alkaline phosphatase (ALP) activity and reduced serum triglyceride levels, indicating a systemic correction of lipid metabolic disturbances frequently observed in ONFH. 108 In addition to bone remodeling, umbilical cord-derived MSCs secrete collagen type VI alpha 2 chain (COL6A2), which activates the FAK/PI3K/AKT(Focal Adhesion Kinase/Phosphatidylinositol 3-Kinase/Protein Kinase B) signaling axis via integrin α1β1 to promote angiogenesis and enhance local vascularization of necrotic regions. 109
A growing body of evidence underscores the importance of the paracrine functions of MSCs, wherein they release a complex array of bioactive molecules—including cytokines, growth factors, chemokines, and extracellular vesicles—that orchestrate local tissue regeneration. Immunomodulatory effects are particularly prominent: MSCs secrete anti-inflammatory cytokines such as interleukin-4 (IL-4) and interleukin-10 (IL-10), thereby dampening the inflammatory milieu and promoting tissue repair.110,111 MSC-conditioned medium has been shown to attenuate femoral head collapse by mitigating cellular senescence, exerting this effect in ONFH models. 112
In summary, MSCs promote ONFH repair through multiple mechanisms, including osteogenesis, angiogenesis, immunomodulation, and inhibition of senescence. These findings provide a robust theoretical and experimental foundation for advancing MSC-based interventions toward clinical application in ONFH therapy.
Exosome-based strategies for the diagnosis and treatment of ONFH
Advancements in biotechnology have shifted therapeutic focus from MSCs themselves to their derivatives, particularly exosomes, in the treatment of ONFH.113,114 In ONFH therapy, MSC-derived exosomes exhibit pleiotropic regenerative effects. The diagnostic and therapeutic roles of exosomes in ONFH are summarized in Figure 4 and Table 4. They promote osteogenesis in hFOB1.19 human osteoblasts by increasing ALP activity and mineralized matrix deposition and upregulating osteogenic genes (osteocalcin [OCN], Runx2, and collagen I). Concurrently, they facilitate angiogenesis in HUVECs, contributing to vascular reconstruction within necrotic tissue. 115 Moreover, M2 macrophage-derived exosomes modulate the immune microenvironment by attenuating neutrophil extracellular trap formation and upregulating VEGF expression, further supporting endothelial angiogenesis and ONFH repair. 116 Exosomes exert multifaceted roles in the treatment of ONFH, including antiapoptotic effects,117–120 promotion of osteogenic differentiation,121–123 and induction of macrophage M2 polarization to ameliorate the immune microenvironment. 124
Despite their high biocompatibility and low immunogenicity, exosome-based therapies face challenges, including limited yield and suboptimal targeting specificity. Their therapeutic potential is highly dependent on the cell source, cellular status, and environmental conditions during culture.125,126 Preconditioning strategies—such as hypoxia—can enhance exosome functionality. 127 For instance, compared with those from normoxic controls, exosomes from hypoxia-conditioned BMSCs significantly increase HUVEC proliferation, migration, and VEGF expression, thereby improving angiogenic outcomes in ONFH. 128 Similarly, pretreatment with atorvastatin enhances the angiogenic capacity of MSC-derived exosomes.129,130
To overcome production limitations, exosome mimetics generated via extrusion techniques have emerged as scalable alternatives with comparable or superior biological activity. 131 For example, simvastatin-preconditioned MSC-derived extracellular vesicle mimetics (SIM-MSC-EM) exhibit enhanced osteogenic and angiogenic activity compared with no treatment, effectively attenuating glucocorticoid-induced ONFH in rat models. Mechanistically, SIM-MSC-EM deliver miR-29b-3p to silence Phosphatase and Tensin Homolog (PTEN), thereby activating the PI3K/AKT signaling pathway to drive bone and vessel regeneration. 132 In addition to their role in therapy, exosomes also play important roles in ONFH pathogenesis and diagnosis.133–136
Despite these advances, several translational challenges remain. Low yield, inadequate bone-targeting ability, and heterogeneity in exosome composition limit clinical application. Future directions include optimizing donor cell preconditioning, engineering surface modifications to enhance bone affinity, and developing synthetic exosome mimetics with tunable properties. Moreover, validation of diagnostic biomarkers such as hsa-miR-200b-3p and hsa-miR-206 in large clinical cohorts could enable early, noninvasive ONFH screening.
In conclusion, exosomes offer a promising multipronged platform for regenerative therapy, early diagnosis, and drug delivery in ONFH. Overcoming current bottlenecks will require the use of integrative strategies spanning bioengineering, molecular diagnostics, and translational medicine to realize their full clinical potential.
Therapeutic potential of apoptotic extracellular vesicles in ONFH
Recent findings highlight that MSCs undergo substantial apoptosis following intra-articular injection, particularly in synovial joints such as the knee. 137 Notably, the apoptotic process itself is now recognized as being biologically active, contributing to tissue repair and immunomodulation. 138 ApoEVs (Apoptotic extracellular vesicles), the extracellular vesicles released during apoptosis, are increasingly recognized as key effectors in this context. 139 Beyond their canonical roles in intercellular material transfer, antigen presentation, and immune suppression, ApoEVs actively participate in tissue remodeling and regeneration, offering new therapeutic opportunities for intractable musculoskeletal disorders such as ONFH.140,141 Experimental evidence supports the role of ApoEVs in promoting osteogenesis. For instance, ApoEVs derived from mature osteoclasts enhance osteogenic differentiation in recipient cells via RANKL-mediated reverse signaling. 142 Systemic administration of exogenous ApoEVs has been shown to maintain MSC homeostasis in pathological environments and mitigate bone loss in murine models, suggesting a protective and regenerative role in ONFH through the augmentation of bone formation capacity. 143
Furthermore, ApoEVs contribute to immune modulation and angiogenesis. Specifically, adipose-derived MSC ApoEVs induce M2 macrophage polarization via the miR-21-5p–mediated suppression of KLF6, concurrently enhancing angiogenic activity.144,145 These findings indicate that ApoEVs exert coordinated effects on osteogenic differentiation, immune regulation, and vascular remodeling—three central processes disrupted in ONFH.
Therapeutic potential of nanozymes in ONFH
ONFH progresses via a cascade of ischemia, oxidative stress, and inflammation, leading to osteocyte apoptosis and impaired bone repair. Nanozymes, a novel class of enzyme-mimicking nanomaterials with high stability, tunable functions, and enhanced physicochemical robustness, have gained great attention in tissue engineering for combating oxidative stress and inflammation. 146
In ONFH, their core therapeutic potential lies in neutralizing excess ROS to alleviate oxidative damage to bone and cartilage tissues; representative examples include Se@SiO2 nanozymes that attenuate methylprednisolone-induced femoral head injury 147 and Mn-Co3O4 nanozymes that protect human MSCs and enhance their osteogenic differentiation, 148 both exerting osteoprotective and regenerative effects. Notably, some nanozymes can even convert ROS into molecular oxygen, ameliorating the hypoxic microenvironment to facilitate angiogenesis and tissue repair. 149 Compared with conventional therapeutics, nanozymes possess synergistic advantages such as targeted delivery, sustained catalytic activity, and potent redox modulation, which perfectly match the complex pathophysiological demands of ONFH, thus emerging as attractive candidates for its precise and localized therapeutic intervention.
However, clinical translation of nanozyme-based strategies still faces challenges, including batch-to-batch variability in manufacturing, insufficient targeting/retention at the femoral head, and the need for rigorous long-term biosafety evaluation of their degradation and metabolic fate in bone tissue.
Polymeric biomaterials for ONFH repair: from structural support to bioactive modulation
Polymeric biomaterials address ONFH challenges by providing structural support and modulating the pathological microenvironment. 150 Natural polymers like chitosan and hyaluronic acid are biocompatible and biodegradable, ideal for scaffolds that support cell adhesion and osteogenic differentiation.151–153 For example, mineralized collagen scaffolds achieve good osseointegration in clinical use. Synthetic polymers such as PCL and PLGA offer tunable mechanics but lack innate bioactivity. 152 Therefore, current research focuses on composite systems that integrate bioactive molecules (e.g., deferoxamine, BMP-2) for controlled release.154–156 These advanced scaffolds simultaneously promote angiogenesis and osteogenesis, as shown in studies where they improved clinical scores and enhanced bone repair in animal models, moving beyond passive filling to active microenvironment regulation. Representative polymeric biomaterials and their therapeutic roles in ONFH are summarized in Table 5.
Organoid and living joint technologies: Emerging frontiers in ONFH regenerative therapy
Organoid technology represents a transformative frontier, using 3D self-organized stem cell constructs to recapitulate native bone-cartilage units. 157 They serve as potent disease models for studying ONFH pathology and screening therapies.158,159 The potential applications of organoid-based regenerative approaches are illustrated in Figure 5. To treat large-scale defects, the field is evolving toward “living biological joints”—bioengineered, integrated replacements with in situ regenerative capacity.160,161 These constructs combine stem cells (e.g., BMSCs), biomaterial scaffolds (e.g., 3D-printed porous titanium with silk fibroin hydrogel), and controlled growth factor delivery. 160 Modular approaches, such as using organoids as “bioblocks” for bioprinting, enable the fabrication of constructs with graded architecture and function.162,163 This integration of technologies heralds a shift from static implants to personalized, biologically active reconstruction for ONFH.
Summary and Prospects
The pathogenesis of ONFH involves an interconnected cascade of vascular injury, hypoxic stress, inflammatory imbalance, and impaired regeneration. Mechanistically, vascular insults create a localized hypoxic microenvironment that triggers oxidative stress and inflammatory cascades, promoting osteocyte death through apoptosis, necroptosis, and ferroptosis. Concurrently, MSC differentiation shifts toward adipogenesis rather than osteogenesis, further compromising bone repair capacity. Traditional treatments offer limited benefits, particularly in advanced disease, driving a paradigm shift toward regenerative strategies along three principal axes: (1) cellular and molecular modulation using MSCs, exosomes, and gene editing; (2) nanobiomaterial systems including antioxidant nanozymes and biofunctional scaffolds; and (3) personalized approaches utilizing organoids and next-generation prostheses.
However, clinical translation faces critical bottlenecks. Vascularization of large—the establishment of functional vascular networks within bulky scaffolds—remains challenging, as current strategies fail to achieve rapid anastomosis with host vessels. Mechanical integration—stable biomechanical interfaces between scaffold and host bone—is essential for weight-bearing sites like the femoral head, yet mismatched mechanical properties often lead to loosening or collapse. Immune compatibility is complicated by allogeneic material-induced inflammation and individual variability in immune phenotypes. Manufacturing reproducibility remains a fundamental obstacle due to donor cell heterogeneity and variable fabrication parameters. Additional translational barriers include scalable manufacturing under Guanosine Monophosphate (GMP) standards, targeting and retention where local delivery (imaging-guided injection, scaffold-based release) and bone-targeting modifications (bisphosphonate conjugation, peptide homing) show promise but face complexity and stability challenges, storage stability requiring cryopreservation, and regulatory standardization lacking unified quality benchmarks. Long-term biosafety also demands rigorous evaluation, as biodegradable polymers must avoid inflammatory degradation products, while inorganic nanomaterials require careful assessment of tissue accumulation and clearance pathways.
Overcoming these barriers will require multidisciplinary integration of advanced biofabrication, immune modulation, standardized quality systems, and harmonized regulatory pathways to enable the successful translation of regenerative therapies for ONFH.
Authors’ Contribution
M.C., B.X., Q.G., and Z.D. conceived the review theme and designed the academic framework. Z.D., L.F., J.W., and Q.W. conducted systematic literature retrieval, screening, and data extraction from databases including PubMed and Web of Science. G.T., Y.Z., S.H., and J.W. performed qualitative synthesis of included literature, evaluated research quality, and analyzed core findings. Z.D., L.F., J.W., and Q.W. wrote the original draft of the article. Z.Z., Z.Z., C.L., X.S., and S.L. reviewed and edited the article for academic rigor, logical coherence, and language accuracy. All authors read and approved the final version of the article and agreed to the submission.
Footnotes
Author Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
This work was supported by the