
Abstract
Background:
Intellectual Disability-Obesity-Brain Malformations-Facial Dysmorphism Syndrome (IDOB-FMD) is a rare genetic disorder characterized by moderate to severe intellectual disability, microcephaly, and brain abnormalities such as a thin corpus callosum, cerebellar hypoplasia, and cerebral white matter hypoplasia, often observed on MRI scans. Additional features include obesity and craniofacial dysmorphism. IDOB-FMD is inherited in an autosomal recessive manner, attributed to Trafficking Protein Particle Complex Subunit 9 (TRAPPC9) deficiency, with a prevalence of less than 1 in 1,000,000. This syndrome shares clinical similarities with Prader–Willi syndrome (PWS).
Results:
In our study, we conducted a clinical evaluation of two Tunisian siblings suspected of having Prader–Willi-like syndrome (PWLS) based on their clinical presentation. Whole-exome sequencing (WES) was employed to identify genetic variants associated with the phenotype. Variant interpretation and segregation analysis were performed to ascertain the pathogenicity of the identified variants. WES revealed a homozygous mutation in the TRAPPC9 gene in both siblings. The variant was predicted to be pathogenic and segregated with the phenotype within the family. Clinical examination was consistent with PWLS, including intellectual disability, microcephaly, and dysmorphism.
Conclusions:
Our findings emphasize the importance of genetic testing in patients presenting with PWLS features, highlighting a TRAPPC9 mutation that expands the known mutational spectrum. This study, the second investigation of PWLS in Tunisia, reports a pathogenic variant in the TRAPPC9 gene underlying the observed phenotype in this family, offering valuable insights into clinical management and genetic counseling. Our identification of this variant not only reveals genetic heterogeneity but also prompts further research to elucidate the genotype-phenotype correlations and molecular mechanisms underlying TRAPPC9-related disorders.
Keywords
Introduction
Prader–Willi-like syndrome (PWLS) is a rare group of genetic disorders that resembles several clinical features of Prader–Willi syndrome (PWS). It is characterized by hypotonia, developmental delays, hyperphagia leading to obesity, and distinctive behavioral issues. However, PWLS differs from PWS in its genetic origins. Unlike PWS, which is usually caused by a specific genetic anomaly in the paternal 15q11-q13 locus, PWLS results from a broader range of genetic alterations, including duplications, insertions, and point mutations affecting various genes (Cheon, 2016; Juriaans et al., 2022).
Genetic variability inherent in PWLS significantly affects its clinical features, leading to a broad range of symptom severity from mild to severe among affected individuals. This variability complicates diagnosis and treatment, emphasizing the need for personalized diagnostic and therapeutic strategies. It highlights the importance of tailored management based on each patient’s unique genetic and clinical profile.
In light of this, managing PWLS requires precise alignment of treatment strategies with the specific genetic abnormalities and clinical features of each case. As of 2023, there has been limited research on PWLS in Tunisia, with only one study reported, revealing a significant gap in our understanding of the genetic landscape and variability of PWLS in the country. This scarcity of data highlights the need for more comprehensive genetic studies and clinical assessments of the Tunisian population. In this context, Trafficking Protein Particle Complex Subunit 9 (TRAPPC9)-related autosomal recessive intellectual developmental disorder has been reported to present with PWLS features, highlighting the overlap between specific gene defects and this phenotypic spectrum.
In this study, we examined the clinical and genetic profiles of two Tunisian siblings initially suspected of PWS. Our findings illuminate the complexities of the disease, revealing a disease-causing mutation in the TRAPPC9 gene linked to this syndrome.
These results support considering the TRAPPC9 gene in the diagnostic evaluation of PWLS phenotypes, especially when routine testing is inconclusive, and highlight its contribution to unresolved neurodevelopmental disorders with overlapping clinical features.
Materials and Methods
Sample collection
Peripheral blood samples were collected from our two patients and their parents in ethylenediaminetetraacetic acid (EDTA) tubes for DNA extraction using the QIAGEN extraction kit and protocol.
Karyotyping
Peripheral blood samples were collected in sterile heparinized tubes for karyotype analysis. Lymphocytes were cultured in Roswell Park Memorial Institute medium (RPMI) medium, supplemented with fetal calf serum, penicillin-streptomycin, and L-glutamine, under sterile conditions in a laminar flow hood. Phytohemagglutinin was added to stimulate cell division, and the cultures were incubated at 37°C with 5% CO2 for 72 h. Metaphase arrest was achieved using colchicine, followed by a hypotonic treatment with 0.075 M potassium chloride (KCl) to improve chromosome spreading. Cells were fixed with a freshly prepared methanol-acetic acid solution, with three washes to ensure purity. Slides were prepared, stained with Giemsa to reveal R-banding patterns, and analyzed using a Cytovision system, with a minimum of 15 metaphase spreads examined per patient.
Fragile X syndrome testing and methylation-specific multiplex ligation-dependent probe amplification
Genomic DNA was extracted, quantified, and aliquoted at a working concentration of 100 ng/µL. For Fragile X testing, polymerase chain reaction (PCR) amplification of the CGG repeat region upstream of the FMR1 gene was performed using a guanine–cytosine (GC)-rich PCR protocol. Amplified products were denatured, combined with the ROX500 size standard, and analyzed by capillary electrophoresis on an AB3500 sequencer. Data were processed using GeneMapper software, and the primer sequences are listed in Table 1.
Primers Used in This Study
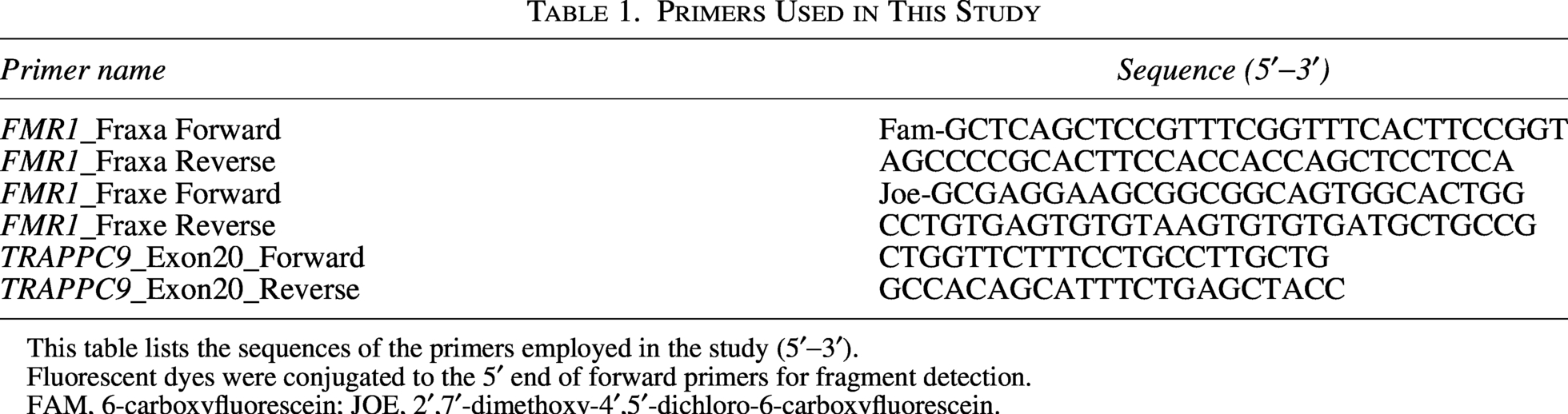
This table lists the sequences of the primers employed in the study (5′−3′).
Fluorescent dyes were conjugated to the 5′ end of forward primers for fragment detection.
FAM, 6-carboxyfluorescein; JOE, 2′,7′-dimethoxy-4′,5′-dichloro-6-carboxyfluorescein.
In parallel, semi-quantitative analyses of copy number and methylation status for imprinted genes located on chromosomes 6 (PLAGL1), 7 (GRB10 and MEST), 11 (H19 and KCNQ1OT1), 14 (DLK1, MEG3, RTL1, and MIR380), and 15 (SNRPN, UBE3A, and NDM) were conducted by Methylation-Specific Multiplex Ligation-Dependent Probe Amplification (MS-MLPA). The ME032 UPD7-UPD14 (48 probes), ME030-C3-BWS-RSS (47 probes), and ME028-B2-PWS-AG (49 probes) probe mix kits (MRC Holland) were used in accordance with the manufacturer’s instructions.
Whole exome sequencing and preliminary analysis
Genomic DNA from peripheral blood samples was used in the whole-exome sequencing (WES) of our two patients. The SureSelect V5-post kit (Agilent TechnologiesTM, Santa Clara, CA, USA) was used to capture genome libraries. Target regions were sequenced on an Illumina HiSeq 4000 system using 150 bp paired-end reads, achieving a depth of 142X (IlluminaTM, San Diego, CA, USA). Alignment of sequences to the human reference genome (GRCh38/hg38) was performed using the Burrows–Wheeler Alignment tool (Langmead et al., 2009). Variant calling was executed using the Genome Analysis Toolkit (GATK v4.2.2.0) (McKenna et al., 2010), and annotation was conducted using SnpEff v4.1 (Cingolani et al., 2012).
Variant filtering followed several steps as follows: (1) prioritization of homozygous variants given the consanguineous family’s history; (2) inclusion of variants with <1% allele frequency within the Genome Aggregation Database (GnomAD v2.1.1; https://gnomad.broadinstitute.org); (3) inclusion of missense, nonsense, and nonsynonymous frameshift variants; and (4) prediction of variant pathogenicity using mutation taster, PolyPhen2, PolyP100way, Combined Annotation Dependent Depletion (CADD), and SIFT scores.
In silico analysis
We checked the remaining variants against Varsome (https://varsome.com/), Gnomad (https://gnomad.broadinstitute.org/), the DISEASES 2.0 database (https://diseases.jensenlab.org/), GeneCards (https://www.genecards.org/), and DisGeNET (https://www.disgenet.org/) for gene-disease associations. Then, we used in silico tools like PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and Mutation Taster (https://www.mutationtaster.org/) to evaluate their pathogenicity and predict their functional impact. Only the TRAPPC9 variant was considered disease-causing in our case.
Sanger sequencing
For verification purposes, the TRAPPC9 variant was validated via Sanger sequencing on the DNA samples obtained from the patients and their parents. Primers are provided in Table 1.
Results
Clinical analysis
In this study, we report two siblings, a 17-year-old boy (P1, III-1) and a 15-year-old girl (III-2; Fig. 1), who presented with facial dysmorphia, intellectual disability with autistic features, speech delay, and delayed growth along with overweight, prompting suspicion of PWS (Table 2). They are the offspring of a Tunisian consanguineous family, with no other individuals exhibiting similar clinical features. However, a history of unspecified psychosis, schizophrenia, and intellectual deficiency was reported in four different members of the extended family (I-6, II-1, II-14, and II-16; Fig. 1).
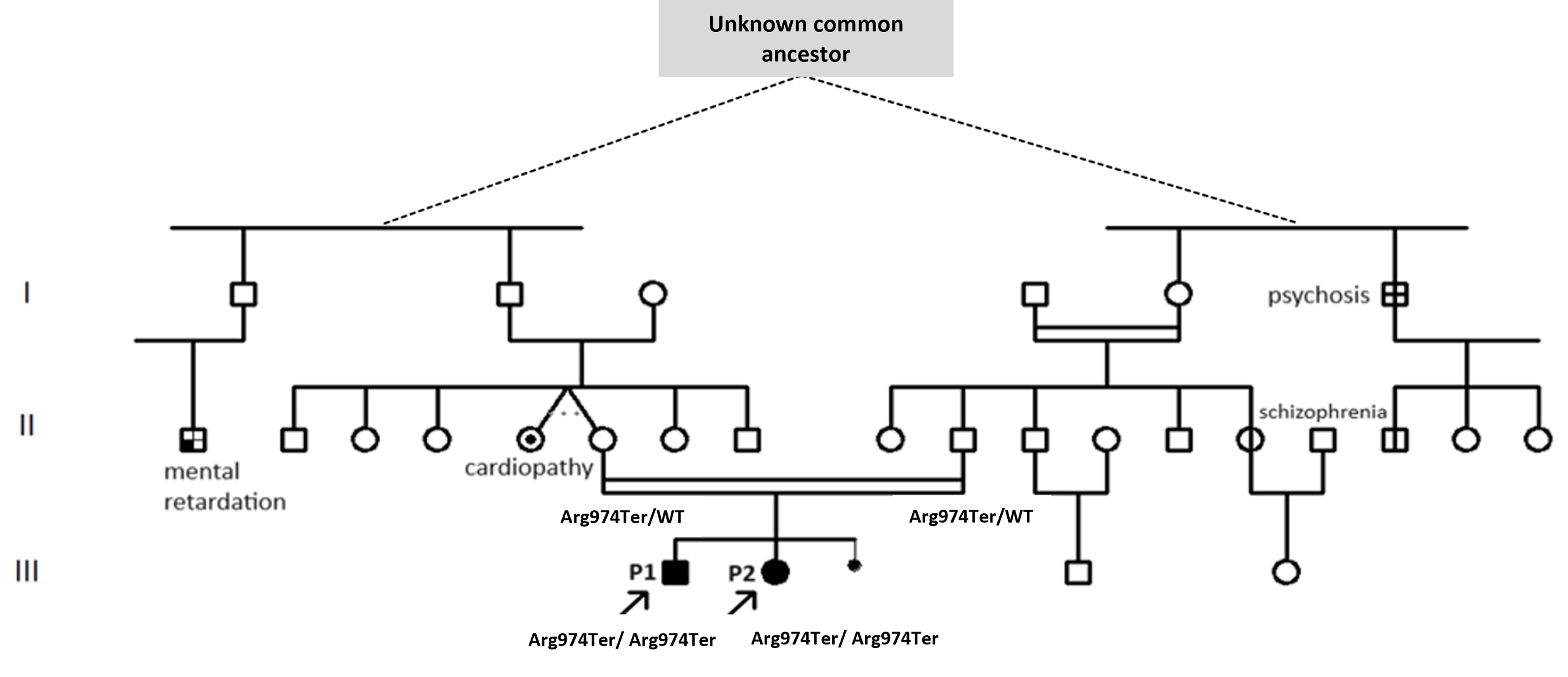
Pedigree of patients P1 and P2’s family representing the family’s medical history. The individuals are distantly related and admitted to being related during the medical interview. Dashed lines denote a presumed relationship via an unidentified common ancestor. Double horizontal lines indicate a consanguineous union. The pedigree illustrates the familial relationships of patients P1 and P2, highlighting the inheritance pattern and relevant clinical conditions observed within the family. Key: Squares represent males and circles represent females. Filled symbols indicate affected individuals, while unfilled symbols indicate unaffected individuals. Arrows indicate the proband individuals (P1 and P2). Genotypes are indicated where available (Arg974Ter/WT: heterozygous; Arg974Ter/Arg974Ter: homozygous variant).
Summary of Clinical Features in Patients P1 and P2

The table presents perinatal history, developmental milestones, growth, feeding, behavioral profile, and neurodevelopmental characteristics for both siblings.
The two patients underwent extensive investigation during prolonged hospitalization in the neuropediatric department, including MRI, standard imaging tests, auditory evoked potential, visual evoked potential, electromyography, pH measurement, and MECP2 gene analysis. The available results are presented in Table 3.
Paraclinical Investigations (Imaging, Metabolic, and Functional Studies)

The table summarizes neuroimaging findings, metabolic and laboratory workup, and gastrointestinal functional assessment.
Genetic analysis
Referred to our Tunisian Genetics Department, both patients underwent genetic counseling and imprinting disorder analysis using the MS-MLPA technique for multi-locus imprinting disorders (Silver–Russell syndrome, transient neonatal diabetes, etc.), Prader–Willi, and Beckwith–Wiedemann syndromes. Results were negative for both syndromes and other methylation abnormalities of imprinted genes on chromosomes 6, 7, 11, 14, and 15. Subsequent evaluations, including conventional cytogenetic analysis, showed normal karyotyping results. PCR amplification of CGG triplets revealed normal FMR1 gene repeats (28 ± 1), with no FMR2 deletion or duplication detected. Both patients underwent WES. Initially, WES analysis identified a total of 35,750 variants. The initial screening prioritized variants within the consanguineous family’s history and with a frequency less than 0.01 in the Genome Aggregation Database (GnomAD v3.1.2; https://gnomad.broadinstitute.org/), resulting in the selection of 46 homozygous variants. Subsequent filtration focused on missense, nonsense, and frameshift variants, yielding a list of 16 homozygous mutations (Table 4).
Remaining Variants After Filtration of Whole-Exome Sequencing Results
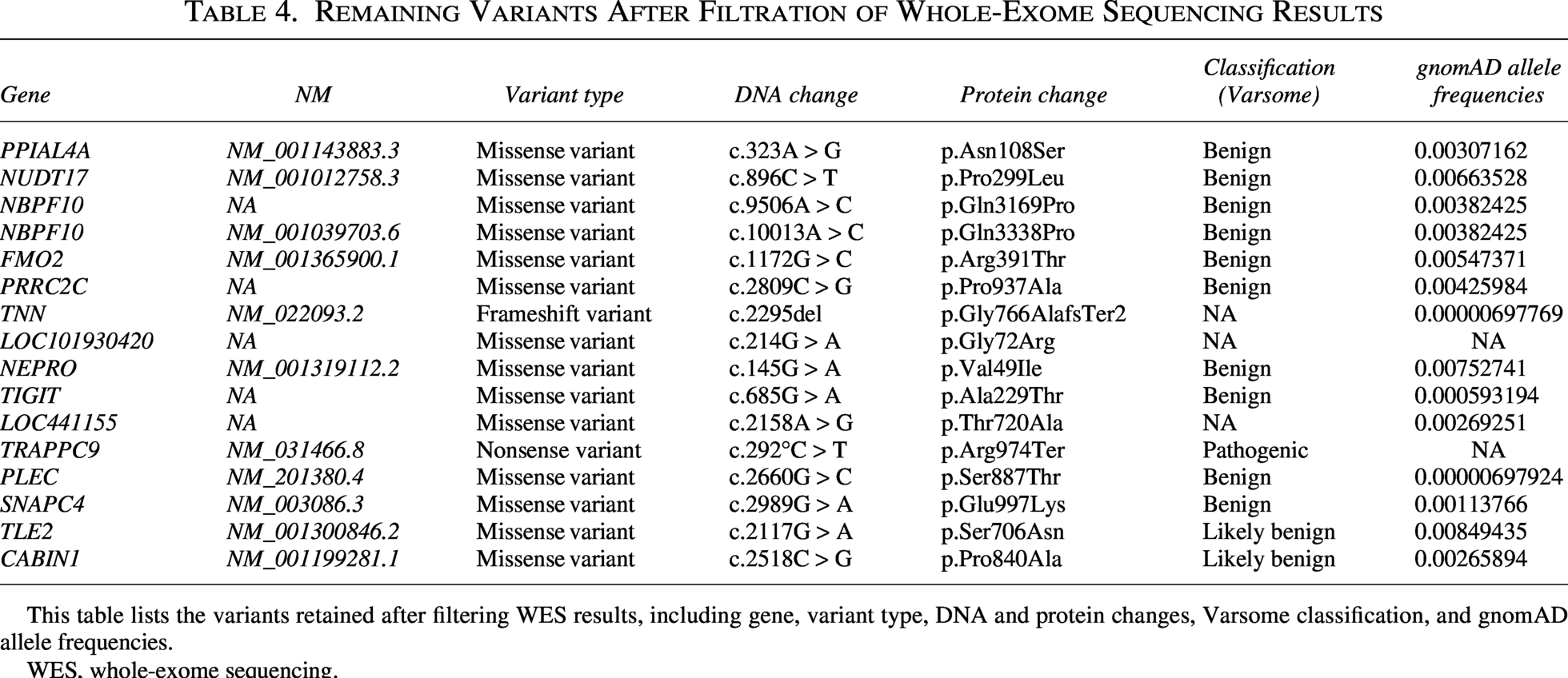
This table lists the variants retained after filtering WES results, including gene, variant type, DNA and protein changes, Varsome classification, and gnomAD allele frequencies.
WES, whole-exome sequencing.
Only a homozygous nonsense variant in the TRAPPC9 gene (c.292°C > T, p.Arg974Ter) was considered as disease-causing variant (Fig. 2). It was confirmed in both patients and their parents (II-6 and II-10) using Sanger sequencing (Fig. 3).
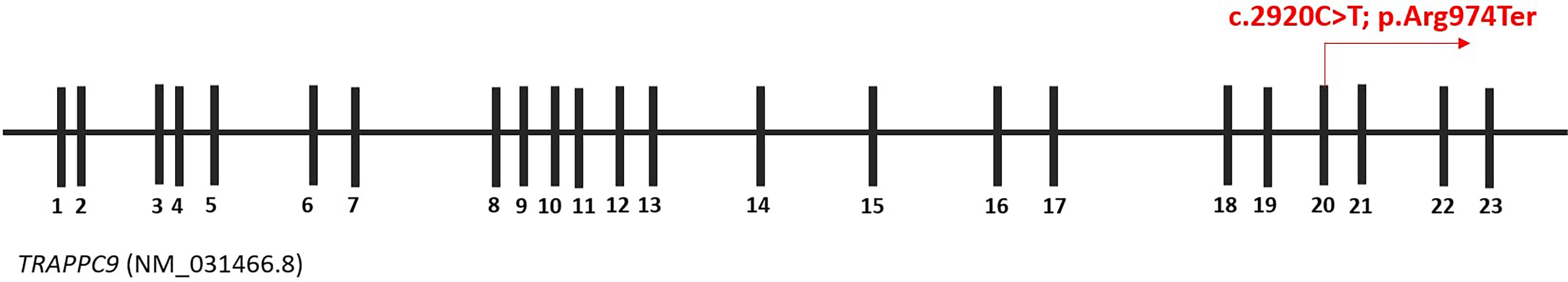
Schematic representation of the TRAPPC9 gene with the identified variant c.292°C > T (p.Arg974Ter). The image provides a schematic representation of the TRAPPC9 gene (NM_031466.8), highlighting the c.292°C > T (p.Arg974Ter) variant in exon 20, which causes a premature stop codon. TRAPPC9, Trafficking Protein Particle Complex Subunit 9.
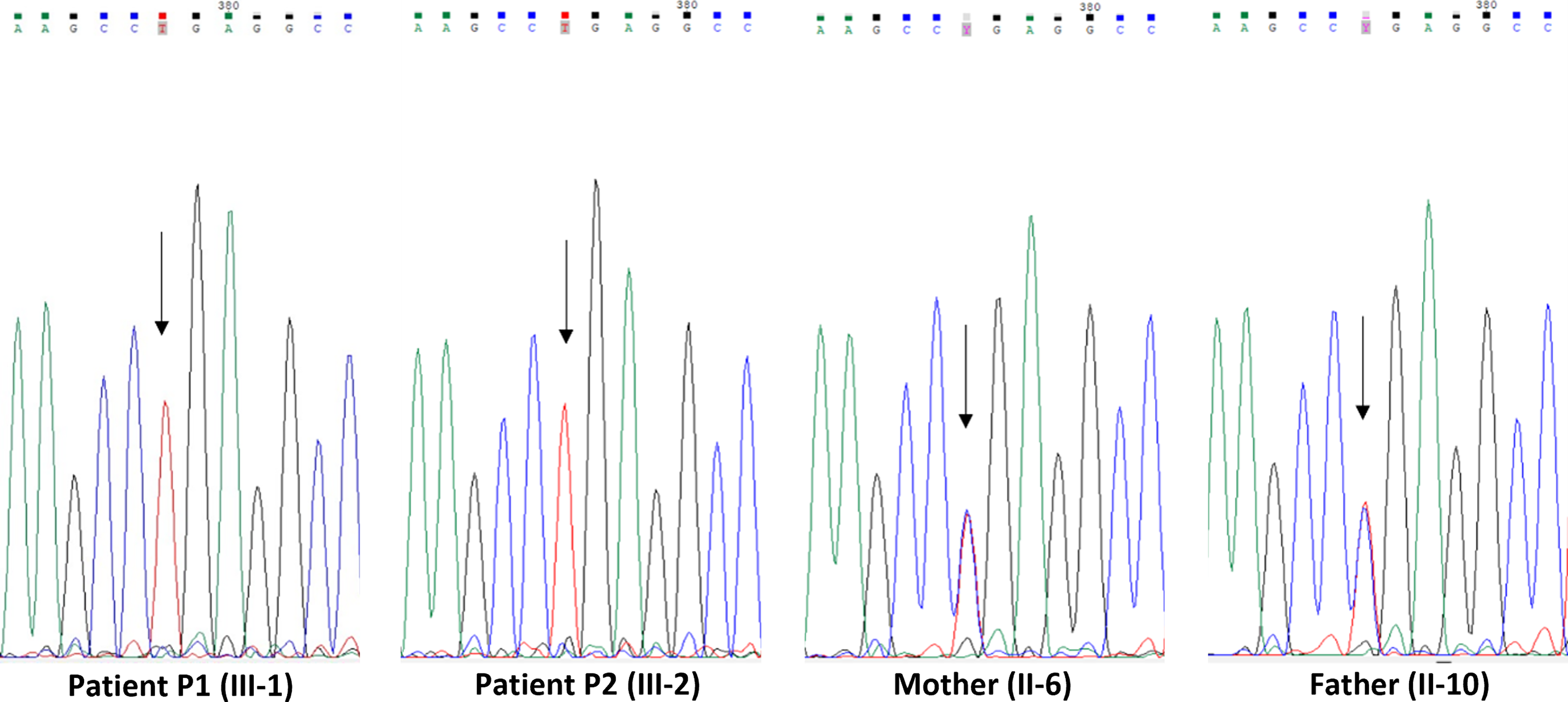
Sanger sequencing electropherograms of the homozygous TRAPPC9 variant c.292°C > T, p.Arg974Ter, in patients P1 and P2 compared to heterozygous carrier parents II-6 and II-10. The electropherograms show the Sanger sequencing results for patients P1 and P2, who are homozygous for the TRAPPC9 variant, and their heterozygous carrier parents (II-6 and II-10). The mutant base is indicated by an arrow.
In silico analysis
In silico analysis using pathogenicity prediction tools indicated a deleterious effect associated with this variant (Table 5). According to the American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines, the c.292°C > T (p.Arg974Ter) variant in TRAPPC9 is classified as pathogenic.
Predicted Impact of Trafficking Protein Particle Complex Subunit 9 Variant by Bioinformatic Tools

This table summarizes the predicted effects of the TRAPPC9 variant as assessed by MutationTaster, Varsome, PolyP100way, CADD, and ClinVar.
TRAPPC9, Trafficking Protein Particle Complex Subunit 9.
Discussion
TRAPPC9 is an essential subunit of the TRAPPII complex and a crucial mediator in vesicular protein transport from the endoplasmic reticulum to the Golgi apparatus. It also interferes with NF-κB activation by interacting with NF-κB-inducing kinase (NIK) and IκB kinase beta (IKKβ) (Alvarez-Mora et al., 2021; Barrowman et al., 2010; Hu et al., 2005; Liang et al., 2020; Zhang et al., 2015). In the context of neurogenesis, TRAPPC9 is important for regulating microRNA-9 expression, which affects neuronal proliferation, differentiation, and migration via nuclear factor kappa B (NF-κB) and cAMP response element-binding protein (CREB) signaling pathways (Yang et al., 2013). C-terminal truncation disrupts the interaction between IKKβ and TRAPPC9 and NIK, prevents recruitment of the IκB kinase (IKK) complex, decreases IκB phosphorylation, and blocks activation of NF-κB (Hu et al., 2005; Zhang et al., 2015). These mutations destabilize TRAPPC9 within the TRAPPII complex, mislocalize IKK vesicles and impair Rab guanosine triphosphatase (GTPase)–mediated trafficking, as well as neurite elongation and branching (Bodnar et al., 2020; Hu et al., 2023; Usman et al., 2022). Loss of C-terminal post-translational motifs further decreases protein stability, as well as cofactor interaction. Consequently, mutations in TRAPPC9 are implicated in various neurodevelopmental disorders, significantly affecting brain development and function (Liang et al., 2020). Full-length TRAPPC9 is required for NF-κB activation and proper neurodevelopment.
The spectrum of TRAPPC9 mutations encompasses a range of genetic alterations, including nonsense mutations, frameshift deletions, splice site mutations, and copy number variations (Hnoonual et al., 2019). Paternal uniparental isodisomy has been identified as a mechanism leading to homozygous deletions in TRAPPC9 (Penon‐Portmann et al., 2023). While nonsense and frameshift mutations resulting in the premature truncation of the TRAPPC9 protein are associated with more severe intellectual disabilities and microcephaly, a clear genotype-phenotype correlation for TRAPPC9 mutations remains elusive. The severity of symptoms varies widely even among individuals carrying the same variant, indicating a high degree of heterogeneity in clinical presentation (Alvarez-Mora et al., 2021; Mir et al., in the TRAPPC9 gene have been linked to Intellectual Disability-Obesity-Brain Malformations-Facial Dysmorphism Syndrome (IDOB-FMD) syndromes (Aslanger et al., 2022; Liang et al., 2020), as reported in the HGMD database (HGMD® gene result [cf.ac.uk]). The identified variant in our study introduces a premature termination codon downstream, leading to the degradation of mutant TRAPPC9 mRNA through nonsense-mediated decay, a cellular mechanism that eliminates mRNAs containing premature stop codons to prevent the production of truncated proteins. This supports the diagnosis of autosomal recessive PWLS due to TRAPPC9 insufficiency.
While both PWLS and IDOB-FMD share overlapping features of obesity, facial dysmorphia, and intellectual disability, they differ in diagnostic criteria, clinical characteristics, and phenotypic spectrum. PWLS is a clinical diagnosis based on physical resemblance to PWS in the absence of chromosomal abnormality at 15q11-q13. It is characterized by hyperphagia, hypotonia, and characteristic neuropsychiatric features. IDOB-FMD is associated with mutations in TRAPPC9 and typically presents with global developmental delay, severe intellectual disability, postnatal microcephaly, dysmorphic facial features, and obesity. However, it lacks the hypothalamic dysfunction and behavioral features seen in true PWLS (Cheon, 2016). Although the clinical presentation in the two affected siblings overlaps with classical PWS, particularly with respect to obesity, hyperphagia, developmental delay, and intellectual disability, molecular tests ruled out any abnormalities within the 15q11–q13 imprinted region. The phenotype observed in this family represents a TRAPPC9-related autosomal recessive intellectual developmental disorder presenting with PWLS features.
Clinical heterogeneity and the broad range of mutations in PWLS syndrome create significant diagnostic challenges. Common symptoms include generalized hypotonia, intellectual disability from mild to severe, global developmental delay, speech impairment, behavioral issues, obesity, microcephaly, and facial dysmorphism such as hypertelorism, synophrys, bitemporal narrowing, a wide nasal bridge, and downturned corners of the mouth with a thin upper lip.
Initial clinical evaluation involves assessing intellectual disability, developmental delay, neurological symptoms, and dysmorphic facial features, with monitoring for associated features like obesity, seizures, and behavioral or psychiatric manifestations. Brain MRI may reveal structural abnormalities linked to TRAPPC9 mutations, such as a thin corpus callosum and cerebellar hypoplasia.
To better delineate the clinical overlap and distinctions between classical PWS and PWLS, we summarized key phenotypic and diagnostic features in Table 6.
Comparison of Prader–Willi Syndrome, Prader–Willi-Like Syndrome, and Trafficking Protein Particle Complex Subunit 9-Related Disorder
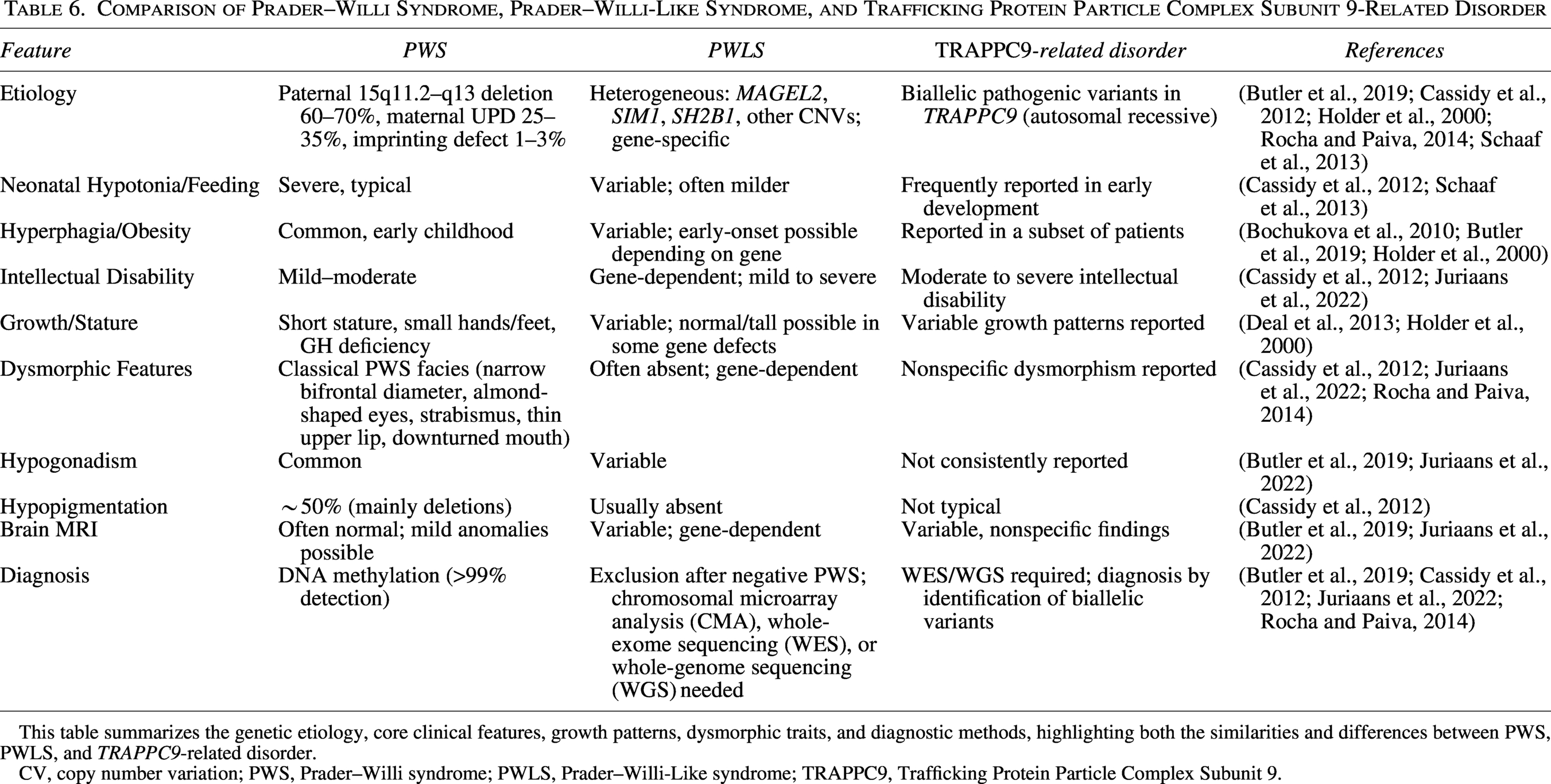
This table summarizes the genetic etiology, core clinical features, growth patterns, dysmorphic traits, and diagnostic methods, highlighting both the similarities and differences between PWS, PWLS, and TRAPPC9-related disorder.
CV, copy number variation; PWS, Prader–Willi syndrome; PWLS, Prader–Willi-Like syndrome; TRAPPC9, Trafficking Protein Particle Complex Subunit 9.
Among the candidate genes identified in the genetic analysis, SNAPC4 was initially considered due to partial phenotypic overlap with neurodevelopmental disorders characterized by global developmental delay and speech impairment. However, our patients lacked the progressive motor deterioration and spastic paraplegia core to the SNAPC4 phenotype (Frost et al., 2023). Instead, the overall genotype–phenotype correlation, characterized by a Prader–Willi-like presentation featuring obesity, delayed growth, and autistic features, strongly favors a TRAPPC9-related syndrome.
In our study, both siblings clinically exhibit features associated with TRAPPC9 loss-of-function mutations, including global developmental delay, severe intellectual disability, postnatal microcephaly, generalized hypotonia, behavioral issues, and obesity. These phenotypes align closely with those reported in other patients carrying other nonsense mutations in TRAPPC9, such as p.R475Ter, which results in comparable neurodevelopmental outcomes (Abbasi et al., 2017). This supports a consistent genotype–phenotype correlation for C-terminal truncating mutations in TRAPPC9 and further substantiates the pathogenicity of the Arg974Ter variant.
Similar pathogenic mutations in TRAPPC9, particularly those introducing premature stop codons or causing frameshifts, have been reported in patients with overlapping neurodevelopmental features such as intellectual disability, postnatal microcephaly, hypotonia, speech delay, and autism spectrum disorder. These mutations have been observed in a variety of populations and genetic backgrounds, highlighting both the recurrent involvement of nonsense and frameshift mutations and the broad phenotypic spectrum observed in TRAPPC9-related neurodevelopmental disorders (Table 7).
Summary of Trafficking Protein Particle Complex Subunit 9 Mutations Reported in Online Mendelian Inheritance in Man (OMIM) and gnomAD Associated with Intellectual Disability
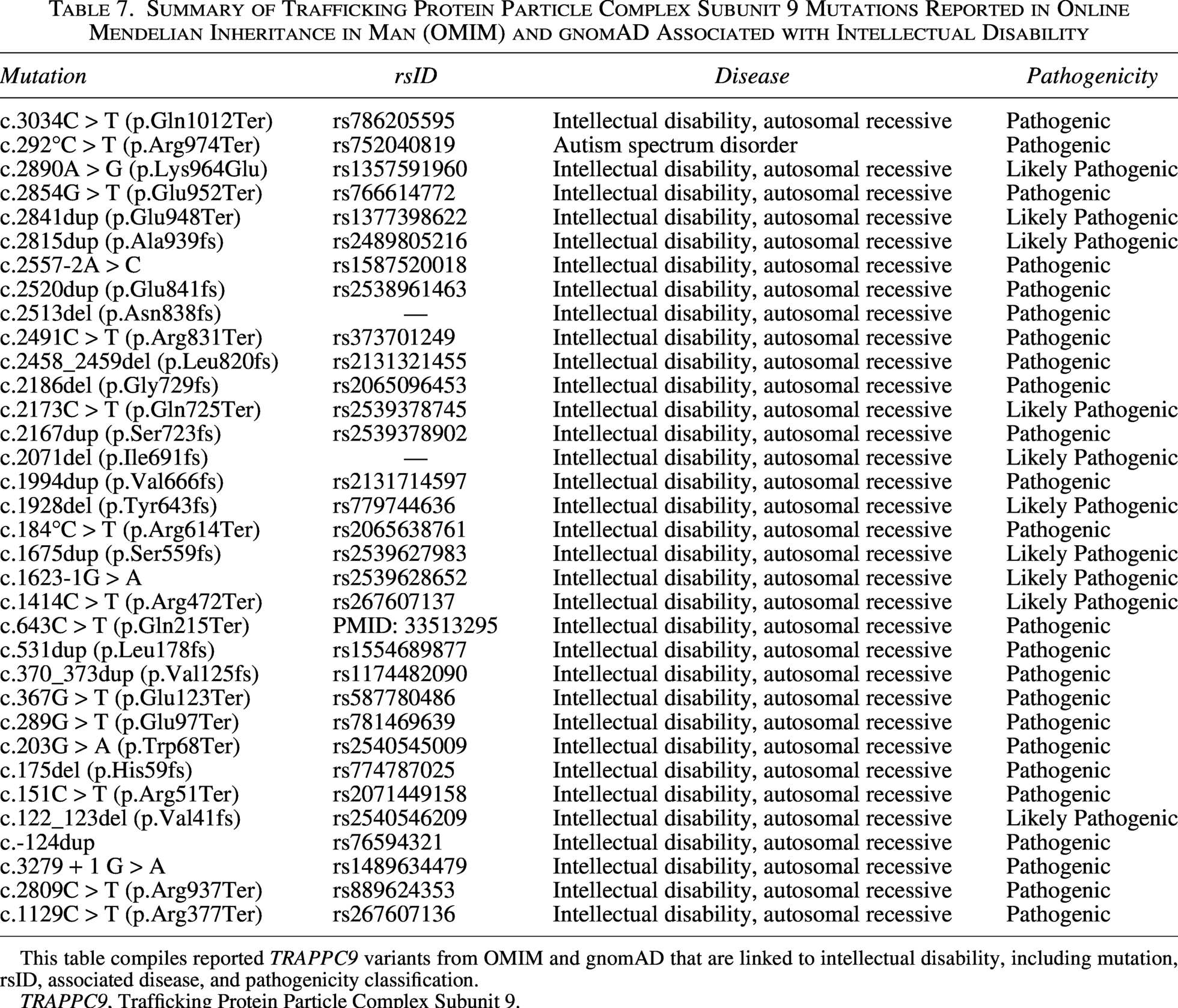
This table compiles reported TRAPPC9 variants from OMIM and gnomAD that are linked to intellectual disability, including mutation, rsID, associated disease, and pathogenicity classification.
TRAPPC9, Trafficking Protein Particle Complex Subunit 9.
While clinical evaluation and imaging studies aid the diagnostic process, genetic testing, especially next-generation sequencing (NGS), offers the most precise evidence for diagnosis. Genetic testing also enables screening of parents for carrier status, informing genetic counseling and family planning decisions. Therefore, genetic testing is crucial in syndrome diagnosis, management, and understanding.
The high prevalence of recessive syndromes in Tunisia is linked to the high rates of consanguineous marriages. Identifying this variant emphasizes the importance of NGS in uncovering rare genetic alterations associated with PWLS.
This report adds further evidence that TRAPPC9-related intellectual developmental disorder extends beyond the classical presentation of autosomal recessive intellectual disability. In our patients, the phenotype included features resembling PWS. This overlap may complicate the clinical diagnosis of syndromic obesity and supports the inclusion of TRAPPC9 in the differential diagnosis of Prader–Willi-like presentations, especially when standard testing for PWS is negative. The recurrence of similar clinical observations in unrelated families suggests that this phenotype is not incidental but may represent part of the broader clinical spectrum associated with TRAPPC9 deficiency. From a practical perspective, these findings reinforce the value of genomic testing in individuals with atypical Prader–Willi-like features, particularly in consanguineous populations where autosomal recessive conditions are more frequent.
Conclusion
Our study adds to the genetic characterization of TRAPPC9-related autosomal recessive intellectual developmental disorder, reporting on a Tunisian family of affected siblings with PWLS features. The finding of a pathogenic TRAPPC9 variant extends the mutational spectrum of this gene and supports genetic heterogeneity in PWLS phenotypes. These observations underscore the significance of testing these patients with syndromic obesity and neurodevelopmental impairment for a broad molecular diagnosis that in turn may guide management, provide accurate prognostication, and lead to appropriate genetic counseling.
However, the study is limited by the single-family sample size. Moreover, further functional validation of this variant and deeper genotype-phenotype correlation studies using broader patient cohorts are warranted to confirm its pathogenic role, clarify phenotypic variability, and refine clinical categorization.
Authors’ Contributions
Conceptualization: M.B.J., H.E.M., D.H., and A.T. Data curation: M.B.J., M.M., and S.N. Formal analysis: M.B.J., A.T., and D.H. Investigation: M.B.J., D.H., A.T., M.M., S.N., I.K., A.R., I.T., and M.G. Supervision: D.H. and A.T. Writing—original draft and writing—review and editing: M.B.J. and H.E.M.
Ethics Statement
This study was conducted in accordance with the Declaration of Helsinki guidelines and regulations. It was approved by the Farhat HACHED University Hospital’s Ethical Committee in Sousse, Tunisia.
Consent
Written informed consent for publication was obtained from the parents. All parents were provided with detailed information regarding the study’s objectives, methods, and potential risks or discomforts associated with participation. To ensure the anonymity and privacy of the participants, biological samples were collected following established ethical guidelines.
Consent for Publication
All authors consent to publication.
Footnotes
Acknowledgments
The authors express our gratitude to the patients and their family members who agreed to participate in this study. They also extend their thanks to all the team members of the Human Genetics and Stem Cell Laboratory at the Research Institute of Sciences and Engineering, University of Sharjah, Sharjah, United Arab Emirates, as well as the Laboratory of Cytogenetics, Molecular Genetics, and Human Reproduction Biology at Farhat Hached University Hospital, Sousse, Tunisia.
Author Disclosure Statement
The authors declare that they have no conflicts of interest.
Funding Information
No funding was received for this study.